The complement system in preeclampsia: a review of its activation and endothelial injury in the triad of COVID-19 infection and HIV-associated preeclampsia
Article information
Abstract
This review assesses the complement system and its activation, with the pathological features of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), human immunodeficiency virus (HIV) infection and preeclampsia (PE). The complement system is the first defensive response by the host innate immune system to viral pathogens, including SARS-CoV-2. SARS-CoV-2 entry results in the release of pro-inflammatory cytokines and chemical mediators to create a “cytokine storm”. Endothelial cell (EC) dysfunction and cell-mediated injury are also present. These factors cause an exacerbated inflammatory state. During HIV infection and PE, various complement components are elevated, causing a hyper-inflammatory state. Furthermore, EC dysfunction and cell-mediated injury are also present. The similarities in pathological aspects of these three disorders may emanate from excessive complement activation. This review serves as a platform for further research on the complement system, coronavirus disease 2019, HIV infection and PE.
Introduction
The complement system is part of the human innate immune response that mediates induction of pro-inflammatory responses, opsonisation of target surfaces, and the lysis of cells and pathogens [1]. Additionally, the complement system clears apoptotic cells, damaged tissue, and immune complexes, thus protecting the host and maintaining homeostasis [2].
Up until 2019, the world’s main public health challenge was the human immunodeficiency virus (HIV) infection, which has a prevalence of 37.6 million [3]. HIV is a retrovirus that impairs cellular immunity, predisposing susceptibility to opportunistic infections [4]. Due to the high prevalence of HIV infection in the South Africa (SA) population, the antenatal prevalence is also very high (30%) [5].
In 2019, a new global health crisis emerged, caused by the novel coronavirus severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). It created medical and economic havoc and devastation worldwide, creating an ongoing syndemic running in parallel with the HIV pandemic [6]. SARS-CoV-2 is an enveloped virus containing a non-segmented, positive-sense ribose nucleic acid molecule, and its mode of transmission is by both droplet and aerosol spread, which are generated during speech, sneezing and coughing within a span of 7-8 meters [7]. Severe SARS-CoV-2 is associated with substantial rates of morbidity and mortality. It is known that SARS-CoV-2 infection causes endothelial injury in response to hyperinflammation and dysregulated immunity [8] and that the complement system and its activation are implicated in the onset of severe infection [9]. Inhibition of the complement cascade may prevent respiratory distress associated with coronavirus disease 2019 (COVID-19) development, particularly in individuals who have a triad of conditions such as chronic hypertension, diabetes and preeclampsia [10,11].
Hypertensive disorders in pregnancy, such as preeclampsia (PE), are associated with high fetal, neonatal, and maternal deaths globally [12]. Endothelial injury is widespread in PE, emanating from oxidative stress and the resultant hypoxia, which predisposes to the release of factors that promote endothelial dysfunction and blood pressure regulation. The pathogenesis of PE remains elusive; however, an exacerbated inflammatory response due to excessive complement activation is evident [13]. Cumulative research evidence links PE with complement dysregulation. Nonetheless, excessive complement activation occurs in the triad of HIV infection, PE and SARS-CoV-2 infection, thereby exacerbating an inflammatory state [14].
This review serves to highlight the complement system in pregnancy, in the triad of pathological complications of PE, comorbid with superimposed by SARS-CoV-2, and HIV infection.
The complement system
Heat labile proteins derived from the liver were discovered in 1891 and named “alexines”. Thereafter, in 1899, Paul Ehrlich renamed them “complement” due to their role in complementing the immune system and antibody function [15]. The complement system is an essential part of the human innate immune response and includes numerous plasma and cell membrane proteins [1]. It is activated by three main pathways: the classical pathway (CP), the lectin pathway (LP), and the alternative pathway (AP), all of which culminate into one terminal pathway (Fig. 1) [16].
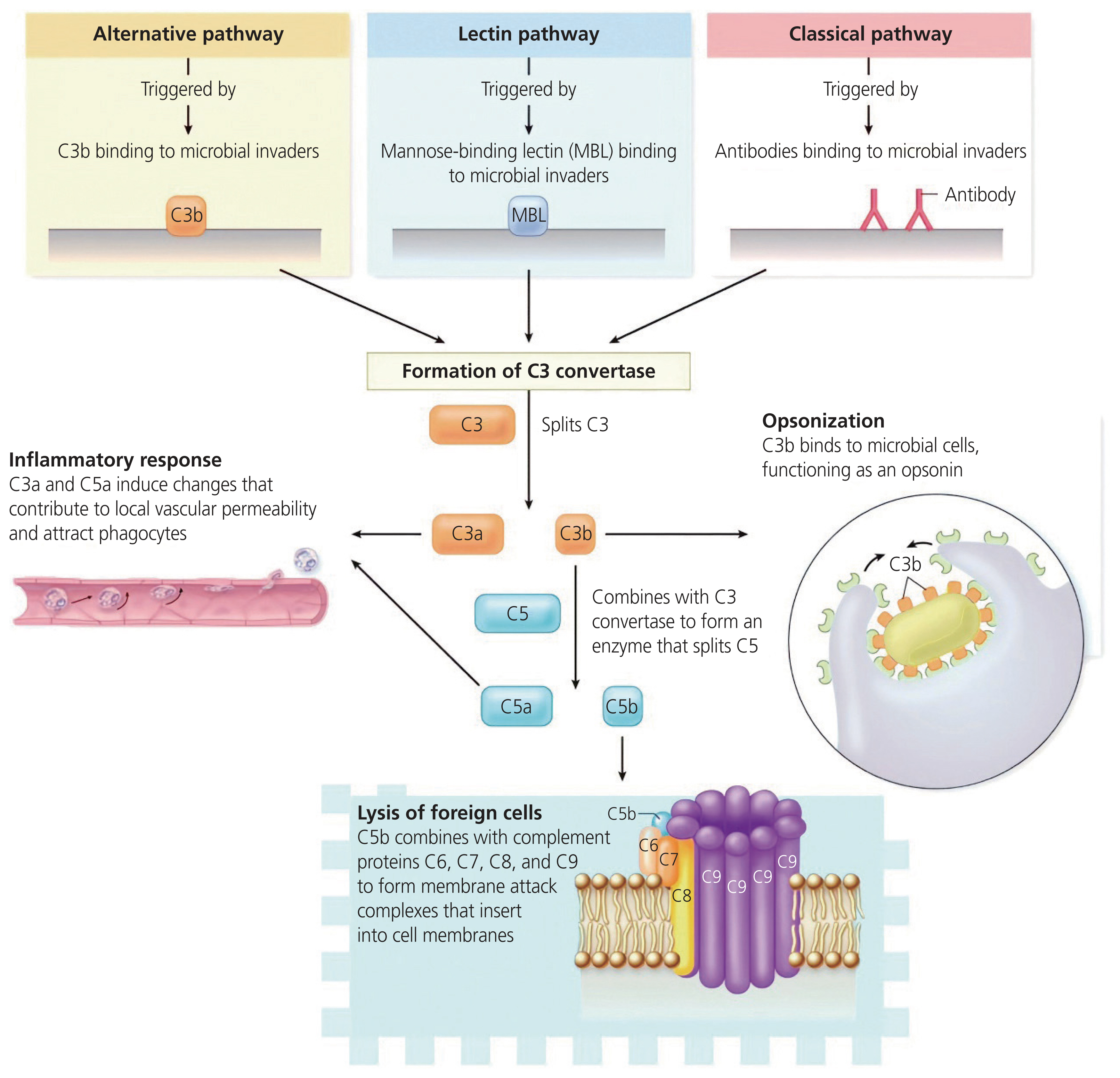
Schematic overview of the complement system, illustrating the three pathways. Activation of the complement cascade occurs via three pathways viz., the classical, lectin, and the alternative pathway. All three pathways result in C3 convertase being produced, which cleaves C3 into C3a and C3b. The complement system opsonises target surfaces, induction of pro-inflammatory responses, and causes lysis of cells and pathogens. Adapted from Beltrame et al. [21].
The CP: is activated due to the binding of recognition molecules C1q to immunoglobulin G (IgG) or immunoglobulin M (IgM) immune complexes bound on the surface of microbes or other structures [17]. This results in a conformational change, causing the activation of C1r and C1s [16]. Thereafter, C1s cleaves C4 and C2 to form a C4bC2A complex, which is a C3 convertase [18].
The LP: is activated by recognition molecules, viz, mannose-binding lectin (MBL), collectin (CL)-10 or CL-L1, CL-11 or CL-K1, and ficolin-1 (M-ficolin), ficolin-2 (L-ficolin), and ficolin-3 (H-ficolin or Hakata antigen) [19].
The AP: is continuously activated at low levels in healthy individuals, where it acts as a “monitoring” system. It is activated by the spontaneous hydrolysis of a thioester bond within C3 to form C3 (H2O), resulting in the binding of factor B and cleavage by factor D to form the C3 convertase C3bBb. The convertase is then stabilised by the plasma protein properdin [20].
PE
Globally, PE is a leading cause of maternal and neonatal morbidity and mortality and is associated with and the development of long-term cardiovascular disease in the mother [22]. Preeclampsia affects approximately 4–10% of all pregnancies [23,24] and is defined clinically by the sudden new-onset of hypertension (blood pressure ≥140 mmHg or ≥90 mmHg diastolic) and laboratory evidence of one more organ involvement (renal, liver, haematological) with or without the presence of proteinuria (≥300 mg or at least two pluses on the urinary dipstick) [25]. During PE, maternal organ dysfunction and severe complications may arise, including hemolysis, elevated liver enzymes, and low platelet count, referred to as the “hemolysis, elevated liver enzymes, and low platelet count” syndrome [26]. There is no cure for PE, except for the early delivery of the fetus and placenta [23]. Clinically, PE may be classified into early-onset (EOPE) and late-onset (LOPE) subtypes. In patients with EOPE, clinical signs and symptoms occur before 33 weeks of gestation, whilst LOPE appears after 34 weeks of gestation [27].
The pathogenesis of PE remains unclear; however, inadequate trophoblast invasion results in a lack of myometrial spiral artery remodelling [28]. This defective remodelling leads to the reduced diameter of spiral arteries and, therefore, a lack of nutrients and oxygen supply to the fetus [29]. This ischemic environment leads to the release of anti-angiogenic factors such as soluble endoglin [23], soluble fms-like tyrosine kinase-1 and inflammatory mediators into the general circulation [30].
HIV infection and highly active anti-retroviral therapy (HAART) in PE
HIV infection remains a global pandemic [3]. South Africa has a high prevalence of HIV infection, 13% (8.02 million), of which 4.7 million are women in their reproductive age [31]. Around 30% of South African women who are pregnant are infected with HIV [5]. Moreover, SA has the most extensive ART rollout globally [3].
Exploiting the life cycle of HIV has led to the development of a combination of at least three anti-retro viral drugs, one of which is HAART [32]. During pregnancy, HAART has the ability to decrease maternal viral replication and prevent mother to child transmission by reducing the plasma viral load in pregnant women [33]. This suggests that HIV infection neutralises the hyper-reactivity of the immune system associated with PE, hence reducing the risk of PE development [34,35]. However, HAART re-establishes the immune response, thus increasing the susceptibility to PE development [36].
Nonetheless, the endothelial damage and vascular dysfunction caused by HIV infection and HAART administration have been reported to predispose patients to develop hypertensive disorders [37].
COVID-19
The emergence of SARS-CoV-2, causing the COVID-19, was declared a pandemic in March 2020 by the World Health Organization. Currently, over 758,390,564 million cases of SARS-CoV-2 have been confirmed, with approximately 6,859,093 million deaths [38].
SARS-CoV-2 enters the human lungs by attaching to the angiotensin-converting enzyme 2 (ACE2) receptor, following priming mediated by transmembrane serine protease 2, present on the alveolar epithelial cells (Figs. 2 and 3) [39,40]. Fever, cough, dyspnea, anosmia, myalgia, or fatigue with evidence of difficulty breathing are common symptoms of SARS-CoV-2 [41,42]. However, the clinical presentation in approximately 80% may be asymptomatic, resulting in mild upper respiratory tract illness [43], but it can rapidly progress to acute respiratory distress syndrome (ARDS), requiring mechanical ventilatory support [44].
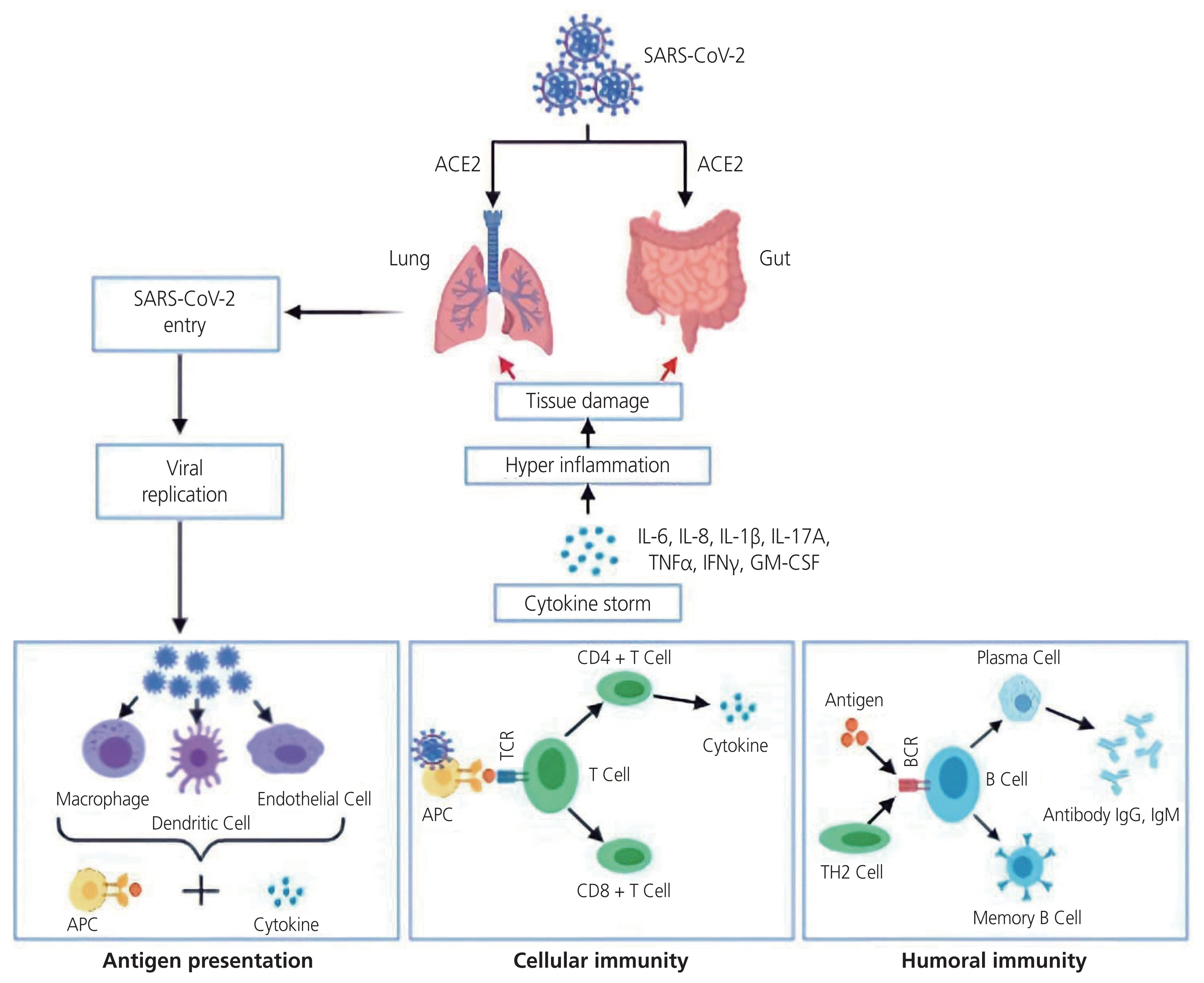
Schematic overview of SARS-CoV-2 and the complement system. SARS-CoV-2 enters via the ACE2 receptor, where it is met by an aggressive inflammatory response caused by the complement system activation. Additionally, cellular and humoral immunity in tandem with antigen presentation becomes activated. This results in large amounts of pro-inflammatory cytokines and mediators being released, consequently exacerbating a hyperinflammatory state and endothelial tissue injury. Adapted from Chatterjee et al. [55]. SARS-CoV-2, syndrome coronavirus-2; ACE, angiotensin-converting enzyme; IL, interleukin; TNF, tumor necrosis factor; IFN, interferon; GM-CSF, granulocyte-macrophage colony-stimulating factor; APC, antigen presenting cell; TCR, T cell receptor; CD, cluster of differentiation; BCR, B cell receptor; TH2, T-helper 2; Ig, immunoglobulin.
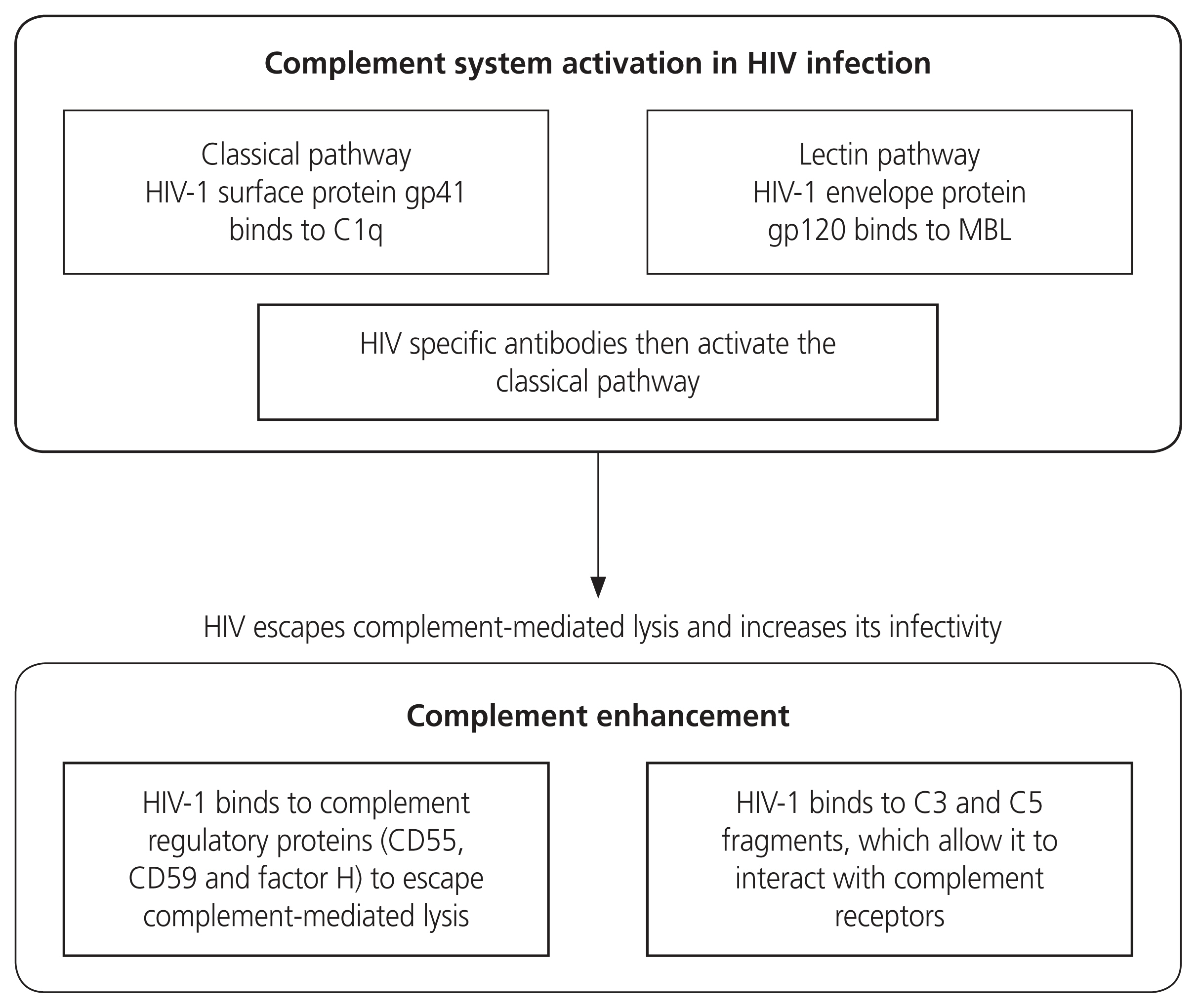
Schematic diagram on the role of the complement system during HIV infection. It is activated by the HIV-1 virus binding to gp41 and gp120. HIV-specific antibodies also contribute to complement activation. HIV-1 uses the regulatory proteins to evade complement-mediated lysis. HIV, human immunodeficiency virus; MBL, mannose binding lectin; CD, cluster of differentiation.
COVID-19 infection is accompanied by an aggressive inflammatory response [45] with the release of a large amount of pro-inflammatory cytokines and chemical mediators in an event known as a “cytokine storm” [46]. This hyper-inflammation is characterised by the infiltration of inflammatory cells in the lungs and other infected organs, particularly in severe forms of the disease [47]. This process is the result of the dysregulated response of the innate immune system via the complement system and is sustained by pro-inflammatory cytokines released by macrophages and other cells at tissue sites [46].
Complement-mediated injury in COVID-19
Evidence of complement-mediated endothelial cell (EC) injury in COVID-19 patients has been observed in multiple organs such as the kidney, lung, liver, and small bowel [48,49]. A study by Bryce et al. [50] noted glomeruli EC with foamy degeneration in the glomeruli of patients with COVID-19. Moreover, endothelial C5b-9 deposition occurs in COVID-19 patients [51]. During SARS-CoV2 viral attachment to ACE-2 receptors on the vascular endothelium, ECs are injured, and complement activation ensues, thereby creating a feedback loop mechanism that induces inflammation [52]. Consequently, elevated von Willebrand factor levels have been reported in patients with severe COVID-19, validating EC injury [53,54].
A study by Huang et al. [46] observed highly elevated levels of plasma pro-inflammatory cytokines (interleukin [IL]-2, IL-6, IL-7, IL-10, granulocyte colony stimulating factor [G-CSF], inducible protein [IP]-10, monocyte chemoattractant protein [MCP]-1, macrophage inflammatory protein [MIP]-1α, and tumor necrosis factor [TNF]-α), known as the “cytokine storm” and this cytokine storm is associated with adverse clinical outcomes, viz, ARDS, organ failure, shock, and even death.
COVID-19 in pregnancy and preeclampsia
Pregnant women may be particularly susceptible to COVID-19 infection due to the physiological and immunological changes of pregnancy [56]. Data on the immune response to SARS-CoV-2 in pregnancy is sparse; however, the timing of COVID-19 infection during pregnancy may cause differences in the maternal immune response, viral clearance, and perinatal outcomes [57]. This is due to the first and third trimesters being pro-inflammatory, which promotes implantation and labour [58]. Pregnant women infected during these trimesters are susceptible to the development of the “cytokine storm”, which predisposes ARDS and subsequent tissue injury [58,59]. Furthermore, high levels of stress and inflammation occur during labour, as well as physiologic changes to a mother’s body after parturition, which may exacerbate poor maternal outcomes [60].
COVID-19 is associated with various systemic complications such as liver injury, renal disease, high blood pressure, and thrombocytopenia [61]. The study by [62] have examined the placentas of COV1D-19 patients and noted a higher incidence of decidual arteriopathy and poor vascular perfusion such as atherosclerosis, mural hypertrophy, and fibroid necrosis; changes that reflect a systemic inflammatory state and hypercoagulability. These findings are similar to the placental changes observed in hypertensive disorders of pregnancy [62]. A study by Estrada-Chiroque et al. [63] reported the first case series of pregnant women infected with COVID-19 and recorded that 20 pregnant women diagnosed with COVID-19 developed PE [64].
One of the defining features of PE is systemic endothelial dysfunction, and this may share a common pathway with COVID-19, as the vascular effects of SARS-CoV-2 include endothelial dysfunction [65]. Mendoza et al. [61] reported that pregnant women with severe COVID-19 have clinical manifestations similar to PE and, with concomitant elevated serum fms-like tyrosine kinase, and decline in placental growth factor. Other studies have also observed SARS-CoV-2 infection creates a pro-inflammatory state, followed by systemic endothelial dysfunction [66,67]. Brosnihan et al. [68] reported that pregnant women with COVID-19 have a higher prevalence of PE. This finding was verified by a recent systematic review that showed COVID-19 infection in pregnancy was associated with a risk of PE development [69].
During normal pregnancy, an increase in the components of the renin-angiotensin-aldosterone-system, including ACE2, occurs [70], which increases the possibility of pregnant women being at a greater risk for SARS-CoV-2 infection. However, in normal pregnancies, a balance between angiotensin II (Ang II) and elevated levels of Ang-(1–7) which maintain vasodilatory responses [71,72]. PE presents an exaggerated Ang II blood pressure response [73]. Additionally, PE is also associated with a decrease in maternal plasma Ang-(1–7) levels [71]. Furthermore, the binding of SARS-CoV-2 to ACE2 causes the downregulation of ACE2 bioavailability and increased Ang II relative to decreased Ang-(1–7), present in PE [74].
SARS-CoV-2 infection mimics microvascular dysfunction, resulting in endotheliitis [75]. Systemic inflammation resulting from vasoconstriction and the resultant ischemia occur [61,76].
The SARS-CoV-2 infection during pregnancy may be prothrombotic, emanating from coagulation abnormalities that result in a hypercoagulable state, which is already exacerbated by PE [77]. Of note, complement activation in COVID-19 comorbid with PE may result in thrombotic vascular injury [78,79]. Furthermore, PE is associated with fibrin deposition in the microvasculature, which causes a compromise in placental perfusion, dysfunction of some maternal organs, and intrauterine fetal growth retardation [80]. Several studies have shown an increase in D-dimer in PE patients [81–83].
The complement system in HIV infection, preeclampsia and COVID-19
During viral infections, the complement system is activated. It prevents the spread of HIV whilst neutralising the virus [84]. Upon entry of HIV-1, the complement system is activated via two HIV glycoproteins, viz, gp41 and gp120 [85]. Thereafter, adaptive immunity is stimulated, activated T cells are synthesised, and the generation of anti-HIV-1 antibodies occurs [86]. During the HIV budding process, HIV-1 virions acquire and bind to complement system regulators such as CD55, CD59, and factor H, which prevents complement-mediated viral lysis [87].
Aberrant complement activation shifts the delicate balance from a normal to a hyperinflammatory state [88]. A study by Perez-Sepulveda et al. [89] suggests that the upregulation of complement components from excessive complement activation results in greater opsonisation of HIV virions. Ito et al. [90] noted the increase of pro-inflammatory cytokines such as IL-6 and TNF-α in the presence of C5a, which promoted HIV-1 infection due to excessive complement activation (Fig. 4).
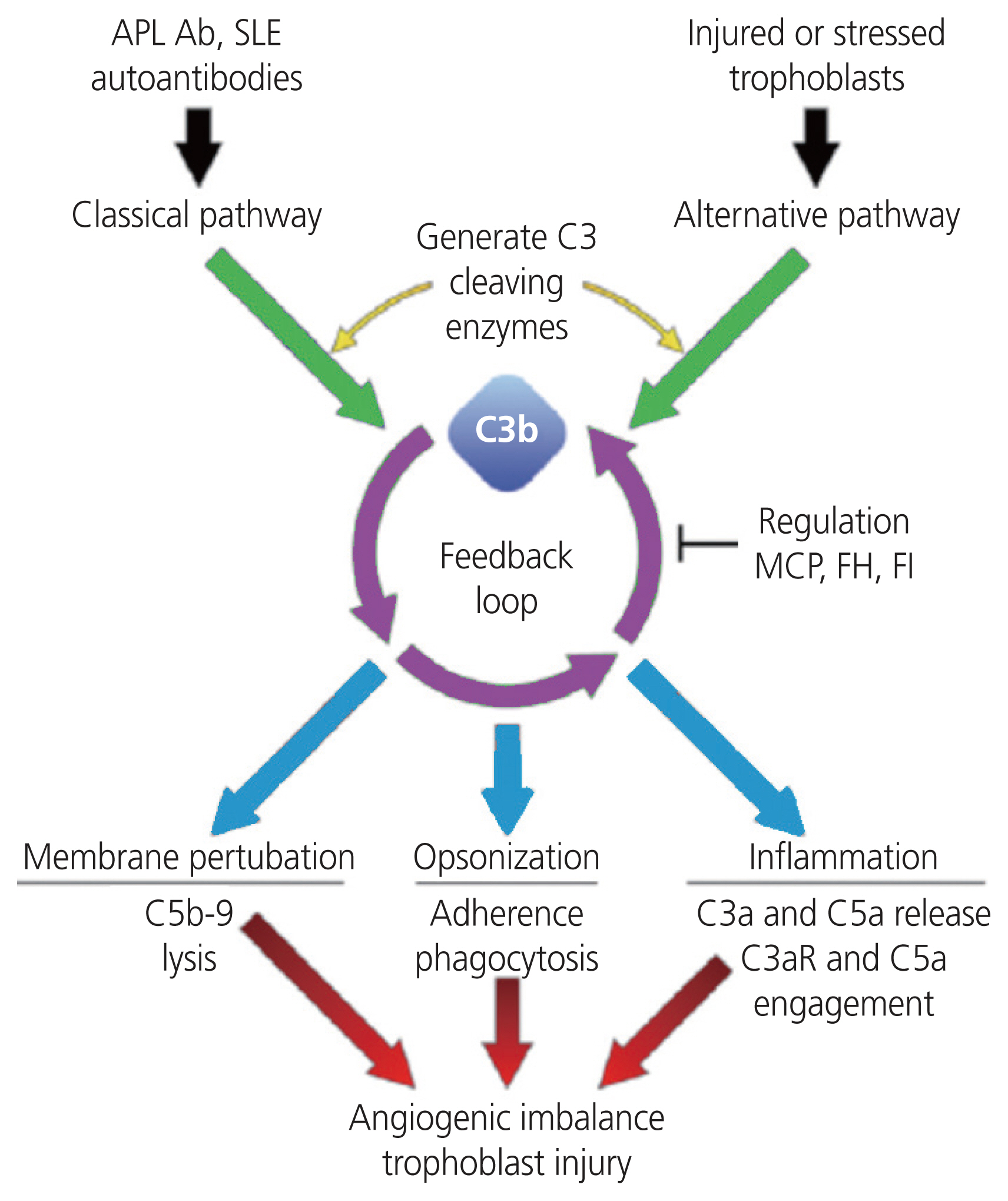
Schematic diagram on the role of the complement system in preeclampsia. The pathogenesis of preeclampsia is not fully elucidated; however, there is a potential link between complement dysregulation and angiogenic imbalance. Complement dysregulation leads to an elevated release of complement components such as C3a, C5a, C5b-9, which cause excessive inflammation, and this can lead to angiogenic imbalances and trophoblast injury. Adapted from Gao et al. [107]. APL Ab, antiphospholipid antibody; SLE, systemic lupus erymatosis; MCP, monocyte chemoattractant protein; FH, factor H; FI, factor 1.
For a successful pregnancy, numerous adaptations of the innate and adaptive immune response occur to support and prevent rejection of the semi-allogenic fetus whilst protecting the mother [91,92]. This is brought about by causing a shift of the maternal response from T-helper 1 (TH1) type to TH2 type immune response, that creates an immuno-tolerant environment [78,93].
In PE, aberrant complement activation results in pro-inflammatory and chemotactic anaphylatoxins [94] (C3a, C4a, C5a, and C5desArg) being released, which cause inflammation vascular leakage, and thrombosis [95]. Moreover, the complement components C3a and C5a are released due to excessive complement activation [96]. They are potent immuno-inflammatory anaphylatoxins, which recruit macrophages, dendritic cells and release pro-inflammatory cytokines, to induce a pro-inflammatory maternal-fetal environment [97]. Aberrant-regulated complement activation can also lead to tissue damage resulting from inflammatory lesions and elevated apoptosis [98]. Furthermore, the synthesis of the membrane attack complex (MAC) leads to calcium influx of target cells, which may cause a metabolic storm, as the sub-lytic MAC induces cell death [99]. The complement system in PE has received considerable attention, with the consensus of an over activation of complement proteins [100,101] and resultant hyper-inflammatory microenvironment [102–104].
Also, in PE, an imbalance of complement activation and regulation may occur due to an incompatibility between the maternal immune system and placental cells [105]. This may initiate local complement activation and early terminal pathway activation, leading to placental damage and ischemia in PE [106].
Notably, an up-regulation of the complement system is crucial for healthy placentation; however, superfluous activation may promote the rejection of fetal-derived tissues. The complement factor 1, complement factor H (CFH), and CFH-related 1 inhibit rejection [107]. Additionally, complement regulatory proteins such as decay-accelerating factor (DAF) (DAF or CD55), membrane cofactor protein (MCP or CD46), and CD59 are expressed by syncytiotrophoblasts to prevent excess complement activation and damage to the placenta [97,101].
Furthermore, elevated complement activation results in products from the CP and LP, viz, C3a, C5a, and MAC being released [18,108]. Yonekura Collier et al. [100] reported significant upregulation of C4d, C3a, and C5b9 in PE compared to normotensive pregnancies. Similarly, Gralinski et al. [106] found increased C4d and C5b9 on the syncytiotrophoblast membranes, whilst Rambaldi et al. [108] reported an increase of complement fragment Bb, a marker of the AP, that precedes the onset of PE.
Recent in vitro and in vitro data implicates complement activation in the severity of both SARS-COV and SARS-COV 2 [109]. Diao et al. [110] described complement activation in SARS-COV using virus-infected C3 mice. Cleaved activation products from C3 such as C3a, C3b, C3c, and C3dg were found in lung tissue as early as the first day of infection. Furthermore, decreased levels of neutrophils, inflammatory monocytes, cytokine, and chemokines occur in C3 deficient mice. These findings suggest that complement activation elevates pathology in COVID-19 infection. Li et al. [109] observed secreted nucleoprotein (N protein) dimers of COVID-19 that activate mannan-binding lectin-associated serine protease 2 (MASP-2), which is the activating enzyme of the LP, leading to the synthesis of C3 convertase and MAC formation. Additionally, an attempt to block MASP-2 and N protein interaction results in lung injury [111]. These findings, together with human proteomic studies [112,113] suggest coronavirus infections and involve multiple complement pathways.
Li et al. [109] found remnants of complement components C3, C4, MAC, and MBL in the alveolar epithelial cells, inflammatory cells, pneumocytes, and alveolar spaces from patients who died of COVID-19. Furthermore, they also reported increased serum C5a levels in the COVID-19 patients. Moreover, Ghai et al. [114] found C5b-9 (MAC) deposition on renal tubules, which indicates the presence of SARS-CoV2 viral infection induced by complement activation in the kidney, and this may contribute to tissue injury and organ dysfunction.
Madhukaran et al. [115] reported increased levels of plasma C5a and C5b-9 were found in moderate and severe COVID-19 patients. Similarly, Clarke et al. [116] found deposits of C5b-9 but also reported C4d and MASP-2 in the microvasculature of patients who died of respiratory failure from septal capillary injury. Moreover, C4d and C5b-9 deposition were localised in the viral S protein terminal of the lung and skin vasculature. Moreover, genetic variants in two of the complement regulators, CD55 and factor H, have been associated with COVID-19 infection, associated with the haploinsufficiency of complement regulatory proteins [117,118].
Complement system changes in COVID-19 and HIV infection during pregnancy
Complement component C1q from the Cp, is a target recognition molecule which links the innate immunity and the adaptive immunity, via binding of IgG and IgM immune complexes [119]. C1q controls antibody-dependant and independent immune function facilitated by cell signalling on effector cell surfaces; and regulates cell differentiation, phagocytosis, cytokine discharge, all of which promote maternal and foetal tolerance during normal pregnancy [120]. Furthermore, it also maintains immune tolerance by means of virus inactivation (promotes the release of pro-inflammatory cytokines) [121]. In the case of a viral infection such as HIV or COVID-19, the complement system will protect the immune system by initiating viral clearance; however, these viruses are able to escape the complement-mediated lysis, thus causing uncontrolled/excessive complement activation and leading to tissue injury, acute and chronic inflammation and coagulation [122]. During pregnancy, the complement system is stimulated to create a suitable micro-environment for both the mother and foetus. The invasion of pathogens such as viruses like HIV and COVID-19, result in the complement system being exacerbated, creating the afore mentioned pathological effects. (Fig. 5).

Schematic diagram of the complement system in HIV infection, Preeclampsia and COVID-19. Excessive or dysregulated complement activation results in the increase of pro-inflammatory cytokines and complement components, causing a hyperinflammatory state. HIV, human immunodeficiency virus; COVID-19, coronavirus disease 2019; IL, interleukin; TNF, tumor necrosis factor; MASP-2, mannan-binding lectin-associated serine protease 2.
In pregnancy, numerous physiological changes of the respiratory and cardiovascular system occur, resulting in pregnant women being vulnerable to pathogen infection. These changes include: an elevated heart rate, decreased lung volume, increased diaphragm, greater oxygen consumption and increased blood volume. Furthermore, the change in long volume and vasodilation causes a resultant airway edema and increased respiratory sections, hence, pregnant women have a deficient tolerance for hypoxia [123]. Moreover, pregnant women have a specific immunological environment which tolerates the semi-allogenic fetus [124]. T cells released by the complement system during pregnancy, have a suppressive effect and results in transient immunosuppression and increasing the susceptibility to virus infection. Additionally, changes in cell-regulated immunity mediated by the complement system, cause the increase in vulnerability of pregnant women towards intracellular pathogens, including viruses such as HIV and COVID-19 [125,126]. During pregnancy, the complement system is activated to some extent to support the development of the placenta, remove apoptotic debris and foetal protection at the maternal-foetal interface, via complement regulators [127]. However, the dysregulation or overactivation may result in tissue damage, release of anaphylatoxin, with subsequent inflammation, thrombosis and vascular leakage, associate with PE [128].
Noteworthy, dysregulation of the complement system emanating from viral infections, has harmful effects for the growth of the neonatal brain [129].
During pregnancy, COVID-19 infection enters the fetal-maternal interface ECs, via the spike protein (S protein), which binds to the ACE2 enezyme. This binding causes competitive inhibition, resulting in ACE downregulation, decreasing Ang 1-7 synthesis, whilst increasing ACE1 activity, elevating Ang II and inducing vasoconstriction, inflammation and vascular permeability [130]. The virus makes its way to the endosome and interacts with toll-like receptors from immune cell, to arouse downstream inflammatory and coagulation pathways [131]. This contributes to the enhancement of an inflammatory response and cell damage both directly and indirectly, as well as decreasing hydroxy ions/carbon monoxide and nitric oxides that aid in vasoconstriction, tissue hypoxia and underperfusion [113]. Additionally, in COVID-19 infected pregnant women, it was discovered that Th17 immunity is greatly increased, resulting in several inflammatory cytokines being released [132,133] and causing a hyperinflammatory state, a hallmark of PE. These inflammatory responses are facilitated by the innate immune system releasing numerous neutrophils, monocytes and macrophages which release different pro-inflammatory cytokines, proteins and molecules accountable for inducing apoptosis and pyroptosis [131].
Simultaneously, the adaptive immune system of the complement system activates the antigen-dependant lymphocytes, via the antigen presenting cells which contain viral antigenic peptides on human leukocytes antigens molecules. This interaction becomes recognized by virus specific T and B cells, Th1 cytokines and Th2 adaptive humoral response by neutralizing antibodies, IgM and IgG [130]. This triggers a hyperimmune response, releasing the “cytokine storm”, as well as other pro-inflammatory proteins such as MCP1, matrix metalloprotease (tissue matrix metalloprotease) or macrophage inflammatory protein 1α. The resultant being increased tissue damage, hypoxia and platelet aggregation hyperactivation [134]. Previous literature has demonstrated dysregulation of the circulatory and placental complement system in PE, suggesting a delicate balance that must be maintained between complement activation and regulation in pregnancy [2,108,135,136].
HIV infection causes hyperactivation of the immune system and chronic inflammation [5]. Notwithstanding, the activation of the complement system is vital in the defence against HIV, however, it can also be a double-edged sword by enhancing HIV infectivity [137]. On the surface of the HIV virus, there are membrane spikes which comprise of 3 glycoprotein 120 molecules joined together and fixed to the transmembrane protein, gp41. This glycoprotein binds to C1q and activates the CP, whilst the HIV-envelope gp120 binds to MBLs and neutralises the virus [133]. The subsequent action being the activation of the complement LP, inhibiting virus entry [138]. Moreover, complement C5a increases TNF-α and IL-6 release, promoting HIV infection and simultaneously inhibiting receptor C5aR, which reverses this action [139]. Furthermore, HIV escapes complement-mediated lysis by binding to the hosts complement regulatory proteins (CD55 and CD59), resulting in the increase of viral infection of T and B cell line [137]. Both PE and HIV are distinguished by chronic inflammation, of which may be exacerbated by excessive or dysregulated complement activation [76].
Complement system changes in normal pregnancy and PE
During normal pregnancy, immune adaptations are made by the complement system to prevent the rejection of the fetus by the mother, whilst protecting against pathogens [77]. The complement system achieves this by causing a shift in the maternal response from Th1 to Th2 type immune response [93]. Trophoblast cells secrete the complement components, C3, C4 and C1q [114]. C1q is synthesised by decidual ECs, implying a correlation between trophoblast cells and spiral artery ECs [120]. Additionally, Madhukaran et al. [115], that C1q plays a role in trophoblast migration and spiral artery remodelling, as C1q deficient mice displayed features of PE such as hypertension and proteinuria.
Harapan et al. [42], observed that pregnant women with increased Bb plasma levels during early pregnancy, are at a higher risk of developing PE later on in pregnancy. This suggests that in PE patients, abnormal complement activation may be present during the first trimester (10–15 weeks). Most studies of complement activation have been focused after the onset of PE and not during the early stages or first trimester [97].
According to Ricklin and Lambris [84], the abnormal regulation of the complement system by the alternative (increased levels of complement factor B and CFH) and classical (elevated C1q) pathways, were observed in the first trimester of patients with PE later in pregnancy. It was reported by Girardi [97] and Lynch et al. [105], that levels of C3a, C5a and sC5b-9 are increased in PE patients, suggesting that abnormal complement regulation plays a role in two stages of PE development [101].
In stage 1 (placenta formation), abnormal complement regulation was found to affect the placental formation, which causes the onset of PE later in pregnancy [98,101]. In stage 2 (after the onset of PE), the complement system becomes activated by local placental ischemia and hypoxia, resulting in a cascade of reactions that contribute to the rapid development of PE [105].
Conclusion
The complement system is the first barrier of defence and is activated in the triad of SARS-CoV-2, HIV infection and PE. The triad results in the dysregulation or excessive complement system activation, the result being the upregulation of various complement components (C3, C3a, C4a/b, C5, C5b-9), pro-inflammatory cytokines and chemical mediators (IL-2, IL-6, IL-7, IL-10, G-CSF, IP-10, MCP-1, MIP-1α, and TNF-α). Therefore, all three conditions represent excessive/dysregulated complement system activation with resultant EC injury and hyperinflammation. However, further research is required to validate these associations.
Notes
Further recommendations
Identification of complement-specific components/pro-inflammatory cytokines and their level of expression in the triad of HIV infection, PE and COVID-19, can potentially serve as biomarkers leading to targeted drug therapy.
Conflict of interest
All authors wish to declare no conflict of interest.
Ethical approval
This study was ethically approved by the Biomedical Research Ethics Committee, UKZN (Ethics number: BREC/0000302 8/2021).
Patient consent
Not applicable, as this is a review paper.
Funding information
Funding for this study was provided by the following people/ organizations: NRF postgraduate scholarship: MND200 708541868 Prof T. Naicker, publication funds UKZN CHS scholarship.