Prenatal diagnosis of a 7q21.13q22.1 deletion detected using high-resolution microarray
Article information
Abstract
We report a case of de novo 7q interstitial deletion detected by conventional karyotyping and by microarray of amniotic fluid sampled during the prenatal period. A 32-year-old pregnant woman was evaluated at our hospital following detection of increased nuchal translucency at 12 weeks and 5 days of gestation. Conventional karyotyping revealed 46,XX,del(7)(q21q22) in 20 interphase mitotic cells, and high-resolution microarray revealed 12.8 Mb (90,625,014-103,430,901) deletion in the region 7q21.13q22.1. Both parents had normal karyotypes. After birth, the neonate displayed several anomalies, including palatine cleft, upslanted and wide palpebral fissure, low-set ears, micrognathia, microcephaly, ventriculomegaly, subglottic tracheal stenosis, hearing loss, and hand/foot deformities, including brachydactyly, polydactyly, and cutaneous syndactyly. This case study helps explain the phenotype-genotype relationship in patients with 7q21.13q22.1 deletion.
Introduction
Chromosome 7q deletion is a relatively rare chromosomal syndrome that is classified by the location of the deleted segment, as follows: proximal (7q11→21), intermediate (7q21→32), or terminal (7q32→qter) [1]. Patients with an intermediate 7q deletion express characteristic prenatal and postnatal phenotypes, including microcephaly, flat nasal bridge, cleft palate, teeth anomalies, cardiac anomalies, ear malformations, facial dysmorphism, micrognathia, genital anomalies, hand/foot malformations, intrauterine growth restriction, glaucoma, hearing loss, feeding problems, mental retardation, and developmental delay. Among these phenotypes, fetal growth restriction and developmental delay/mental retardation are the most common findings, whereas hand/foot malformations, especially ectrodactyly, are the most specific physical findings [2].
Here, we report a case of de novo 7q21.13q22.1 deletion diagnosed at 17 weeks of gestation using conventional karyotyping and high-resolution microarray. Most previous case studies involving 7q21.13q22.1 described a broader region or lacked precise breakpoints because they were analyzed before high-resolution microarray became available. In this report, we describe the 7q21.13q22.1 deletion phenotype in detail and review previous studies regarding cases with similar chromosomal breakpoints and genes mapped to that region.
Case report
A 32-year-old pregnant woman presented to our hospital at 12 weeks and 5 days of gestation for evaluation following detection of increased fetal nuchal translucency (NT). Her delivery history was G0P0A0. She and her 34-year-old husband had no family histories of congenital disorders or malformations. Prenatal maternal serum screening was normal, and prenatal fluorescence in situ hybridization, using probes of X, Y, and CEP 8 (Abbott Laboratories, North Chicago, IL, USA) according to the manufacturer's instructions, was unremarkable. Conventional G-banding karyotyping was performed on metaphase mitotic cells obtained from amniotic fluid, according to standard procedures. The results indicated a karyotype of 46,XX,del(7)(q21q22) (Fig. 1A). Karyotyping results from both parents were normal. To delineate the precise breakpoint, genome-wide high-resolution microarray (Affymetrix Cytoscan 750K array, Affymetrix Inc., Santa Clara, CA, USA) was performed. A hemizygous deletion on 7q21.13q22.1, covering 90,625,014-103,430,901 bp region (Fig. 1B), was detected. The final karyotype was concluded as 46,XX,del(7)(q21.13q22.1).
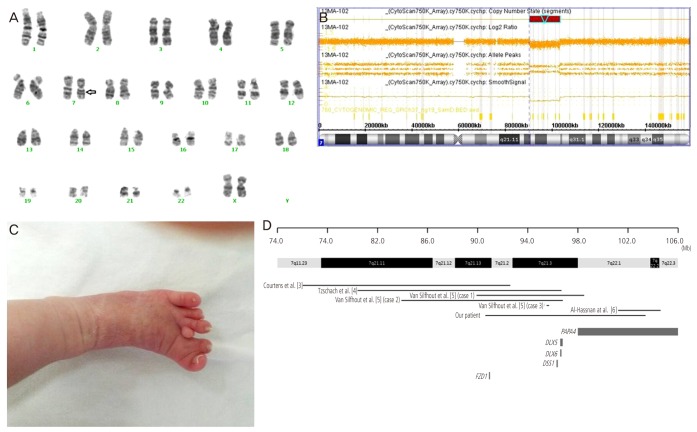
(A) Karyogram obtained prenatally from sampled amniotic fluid. Intermediate deletion of chromosome 7 between bands 21 and 22 was observed (arrow indicates the deleted segment). (B) The microarray result of the patient. The result showed a log reduction and loss of heterozygosity at the 7q21.13q22.1 region, which had a maximal size of 12.8 Mb from 90.63 to 103.43 Mb of chromosome 7. (C) Polydactyly between the second and third toes of the infant's left foot. Cutaneous syndactyly of the first and second toes; retroflexed first and fifth toes are observed. (D) Overview of the chromosome 7q21-q22 region, showing genes related to the clinical phenotype of 7q21q22 deletion. Different phenotype results among previous cases and ours suggest the existence of other factors, besides DLX5, DLX6, and DSS1, affecting the expression of split hand or foot. And the unique chromosomal region deleted in our patient, which does not fully overlap with deletions harbored by other cases, will be applicable to functional studies of the 7q22.1 region [3,4,5,6].
Fetal magnetic resonance imaging and ultrasonography (USG) were performed at 27 weeks and 3 days of gestation. No phenotypic abnormalities were detected except for increased NT to 4.5 mm. Mild polyhydroamnios was observed.
At 36 weeks and 5 days of gestation, the female fetus was delivered by caesarean section due to fetal distress. At birth, she exhibited poor-to-moderate crying and activity, and her 1- and 5-minute Apgar scores were 6 and 6, respectively. The neonate weighed 1,730 g (<3rd percentile), was 38 cm long (<3rd percentile), and had a head circumference of 30 cm (<3rd percentile). Laboratory tests including blood cell count, liver function test and arterial blood gas analysis were unremarkable. The following abnormalities were observed upon physical examination: low-set ears, ear malformation, palatine cleft, upslanted and wide palpebral fissure, micrognathia, microcephaly, short fingers on both hands, polydactyly between the second and third toes, cutaneous syndactyly of the first and second toes, and deformities of the first and fifth toes of the left foot (Fig. 1C).
Serial head USG indicated asymmetric lateral ventriculomegaly, and transthoracic echocardiogram revealed a 2.5 cm patent ductus arteriosus (PDA) and a 2.4 cm atrial septal defect (ASD). Computed tomography scans of the neck and chest revealed subglottic tracheal stenosis and persistent left-sided superior vena cava (LSVC). Abdominal USG was unremarkable. Visual and audiologic examinations were not conducted due to the infant's young age, but auditory brainstem response testing indicated probable hearing loss. Repeat karyotyping using the infant's peripheral blood indicated a karyotype of 46,XX,del(7)(q21.13q22.1), identical to the results obtained from prenatal amniotic fluid sampling.
At manuscript submission, the infant had undergone PDA ligation surgery and was being treated in the intensive care unit of our hospital with oxygen supplementation, blood transfusion, and antibiotics due to repeated respiratory failure and infections.
Discussion
Although several 7q deletion cases involving 7q21.13q22.1 have been reported, most of them involved a broader region than 7q21.13q22.1, and only a few cases were able to delineate the precise chromosomal breakpoint by high-resolution microarray analysis (Table 1) [3,4,5,6]. Since the emergence of high-resolution cytogenetic techniques in clinical diagnostics, the identification of more detailed breakpoints and the subsequent reestablishment of phenotype-genotype correlations become possible. Moreover, high-resolution detection methods have facilitated the discovery of genes related to specific phenotypes [6].

Clinical manifestation of patients with a similar deletion region with of our patient
We attempted to compare our case with earlier reports describing a similar breakpoint (Fig. 1D) [3,4,5,6]. The phenotypic findings of our case were similar to previous cases and included prenatal growth retardation, facial dysplasia, and hand/foot malformations (Table 1). However, the most specific finding from previous reports, ectrodactyly, was not found in our patient. Ectrodactyly (split hand, cleft hand, or lobster claw hand) describes the deficiency or absence of one or more central digits of the hand or foot and is also known as split hand/foot malformation (SHFM). SHFM is a heterogeneous disorder that can occur in isolated nonsyndromic form or as one phenotype of several syndromes. To date, six different forms of nonsyndromic SHFM, deemed SHFM1-6, have been described in humans [7]. Of these, SHFM1 maps to 7q21q22, a region that spans several candidate genes, such as SHFM type 1 (SHFM1), distal-less homeobox 5 (DLX5), and distal-less homeobox 6 (DLX6) [8]. Mice expressing a double knockout of DLX5 and DLX6 showed typical ectrodactyly, and the SHFM1 gene is expressed in developing limb buds and is thought to be involved in the pathogenesis of SHFM [9].
Most, but not all, patients harboring a deletion or rearrangement in these candidate genes express some type of SHFM. Tzschach et al. [5] reported a case involving deletions in SHFM1, DLX5, and DLX6, but without SHFM. These authors suggested the 300 kb region distal to the SHFM1, DLX5, and DLX6 genes and between BAC RP11-800O14 and marker D7S618 as a causative defect region for SHFM. However, a report by van Silfhout et al. [7] (case 3) involving inv(7)(p22q21.3) showed typical SHFM without mutations in SHFM1, DLX5, or DLX6. Our case and another case by van Silfhout et al. [5] (case 2) harbored deletions spanning the three candidate genes and the above regions suggested by Tzschach et al. [4], but they did not exhibit distinct ectrodactyly. Instead, our case expressed brachydactyly of the hand, cutaneous syndactyly, and central polydactyly of the foot. This discrepancy could be explained by reduced penetrance of SHFM1, DLX5, and DLX6, or by an insufficiency in the development of ectrodactyly due to haploinsufficiency of the causative genes. Bernardini et al. [10] proposed an alternative candidate gene, FZD1, associated with SHFM1 in a patient with microdeletion of 7q21.13. FZD1 is a member of the "frizzled" gene family, and the FZD1 protein is activated by Wnt1 and Wnt3A, which interact with DLX5 and DLX6. Additional cases describing detailed deletions in this region might contribute to a more precise characterization of the SHFM locus.
Our case displayed brachydactyly of the hand, cutaneous syndactyly, and central polydactyly of the foot. Although the polydactyly could be associated with SHFM, it is a rare phenotype in patients with the 7q21q22 deletion. Based on the locations of the extra digits, polydactyly can be classified as preaxial, involving the thumb or great toe; postaxial, affecting the fifth digit; or central, involving the 3 central digits. Our patient exhibited central polydactyly for which an associated gene has not been reported to date. However, PAPA4, one of the genes related to postaxial polydactyly type A, has been mapped to 7q21-34, which spans the deleted region in our case [11].
Hearing loss is a common characteristic of patients with SHFM1, and it may be accompanied by Mondini dysplasia of the inner ear [12]. It is not known whether SHFM and hearing loss are caused by mutations in the same gene or different genes in the SHFM1 region [4]. Notably, autosomal-recessive SHFM1 accompanied by hearing loss, termed split hand/foot malformation 1 with sensorineural hearing loss, is associated with a mutation in DLX5 [13].
PDA, ASD, and persistent LSVC are rare features in 7q21q22 deletion cases, but these cardiovascular anomalies sometimes are observed in other premature or intrauterine growth restriction infants, so the phenotype-genotype relation is not clear [14]. Prior to this case study, subglottic tracheal stenosis had never been reported in a patient with intermediate del(7q), and no genes or chromosomal abnormalities on 7q have been associated with tracheal stenosis to date.
The phenotype of 7q deletion syndrome is heterogeneous and nonspecific. Therefore, early detection of 7q deletion using only clinical manifestations or prenatal diagnosis is very difficult. Our patient displayed increased NT, but other prenatal screening tests were within normal ranges. Although a previous case study associated 7q deletion with abnormal triple screen results in the prenatal period, most 7q deletion cases yield normal prenatal screening results [15]. Increased NT is a commonly observed, nonspecific feature that can be associated with many chromosomal abnormalities, but it could be a useful indicator of 7q deletion, as in our case.
In brief, we describe a case of 7q21.13q22.1 deletion identified following an abnormal NT result and confirmed by karyotyping. The precise chromosomal breakpoint was delineated by high-resolution microarray. Our case involved deletions of SHFM1, DLX5, and DLX6, which are candidate genes for SHFM; however, our case lacked distinct SHFM, instead displaying polydactyly. This case report could be applicable to studies of phenotype-genotype relationships in patients with intermediate 7q deletion, particularly those with SHFM.
Notes
No potential conflict of interest relevant to this article was reported.