


Introduction
A congenital cystic adenomatoid malformation (CCAM) is a cystic mass that is histologically characterized by abnormal proliferation of the terminal respiratory bronchioles [1], and rarely occurs, with a considered incidence of 1:25,000 to 1:35,000 pregnancies [2]. CCAM usually involves only one lobe of the lung and cystic structures arise from an overgrowth of the terminal bronchioles with a reduction in the number of alveoli [3]. In the normal lung, the anatomical development periods have been divided into five distinct phases: embryonic (3-7 weeks), pseudoglandular (7-17 weeks), canalicular (17-29 weeks), saccular (24-36 weeks), and alveolar (36 weeks to maturity) [4]. Most instances of CCAM arise during the pseudoglandular period when a rapid expansion of the conducting airways and peripheral lung tubules occurs.
CCAM is prenatally classified as macrocystic or microcystic, based on its in appearance on a prenatal ultrasound [4]. Microcystic CCAM, which appears as solid echogenic mass, tends to regress spontaneously after approximately 26 to 28 weeks of gestation [5]. On the other hand, macrocystic lesions, which contain single or multiple cysts that are 5 mm or larger in diameter, generally do not regress as fluid accumulates within the cysts [6]. Macrocystic CCAM also carries a significant risk of pulmonary hypoplasia, pleural effusion, fetal hydrops, and subsequent fetal demise [4].
The prenatal therapies in current practice for these lesions include aspiration of the cyst, thoracoamniotic shunt, injection of sclerosing agent, and open fetal surgery [7,8,9]. The aim of our study was to introduce our experiences using various modalities to treat fetuses diagnosed with macrocystic CCAM in a single Korean center.
Materials and methods
Following institutional review board approval, we retrospectively analyzed fetuses with macrocystic CCAM who underwent intrauterine therapy between August 1999 and January 2012 at the Asan Medical Center, Seoul, Korea. Among 319 fetuses diagnosed with CCAM by prenatal ultrasound, 129 were postnatally confirmed with CCAM. Of 25 neonates confirmed with macrocystic CCAM, six underwent intrauterine treatment, and these cases were finally analyzed (Fig. 1). The following parameters were assessed: the gestational age at diagnosis, the size of largest cyst within the CCAM, the presence of mediastinal shifting, any associated polyhydramnios, the presence of pleural effusions, ascites or fetal hydrops, and any combined anomalies. From 2011, we also assessed fetal left cardiac function in CCAM cases by measuring the modified myocardial performance index (Mod-MPI). Indications for prenatal intervention included a largest cyst more than 2 cm, and the presence of mediastinal shifting or fetal hydrops. In utero treatment modalities were as follows: aspiration of cysts, thoracoamniotic shunt, and injection of the sclerosing agent. OK-432 (Picibanil, Chugai Pharmaceutical Co., Tokyo, Japan), a lyophilized incubation mixture of group A Streptococcus pyogenes, was used as the sclerosing agent. OK-432 at a concentration of 1 Klinische Einheit (KE) in 10 mL normal saline was injected into the macrocystic cavity. The delivery characteristics that were recorded included the gestational age at delivery, birth weight, and neonatal outcomes, such as the Apgar score, and the presence of respiratory complications. A diagnosis of CCAM was confirmed in all neonates by postnatal chest computed tomography and postoperative pathology.
Results
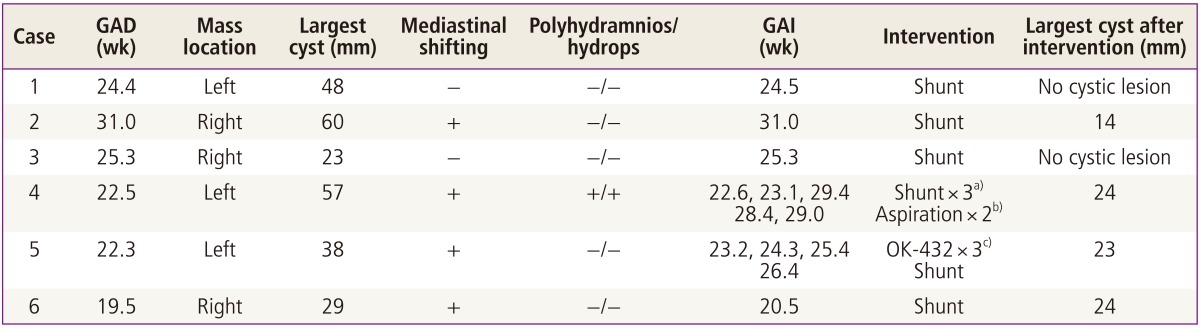
Table 1 demonstrates the clinical characteristics of the six fetuses with macrocystic CCAM analyzed in this report. The median gestational age at diagnosis was 23.5 weeks (range, 19.5-31.0 weeks), and the median gestational age at first clinical intervention was 24.0 weeks (range, 20.5-31.0 weeks). The mean size of the largest cyst within the CCAM at the initial assessment was 42.5 ±15 mm. Four of these cases were associated with mediastinal shifting, and one also showed fetal hydrops. There were no chromosomal or combined anomalies detected in any of the fetuses.
All fetuses underwent shunting within the cysts, which almost eliminated the cystic lesions in three fetuses (cases 1, 2, and 3). One fetus (case 4) required repeated shunting with aspiration of the cysts because multiple large cysts had presented within the mass. After the second shunting procedure within each of the two large cysts, the fetal hydrops improved. However six weeks after the initial shunting, a new 4 cm sized cystic mass developed and fetal hydrops reemerged. Despite a second series of aspirations of these cysts, the hydrops was not again alleviated and shunting was therefore performed again at 29.4 weeks of gestation. Following these procedures, only small cysts persisted until birth.
One of our six cases was treated with OK-432 (case 5) (Fig. 2). An aspiration of 45 mL of cystic fluid followed by injection of 0.07 KE (0.7 mL) was performed at 23.2 weeks of gestation. This procedure was repeated twice with increased does of OK-432 (0.12 and 0.2 KE, respectively). Although there was a subsequent marked reduction in the size of the cyst after each injection of OK-432, the fluid was found to have reaccumulated at the 1 week follow-up. Therefore shunting was performed at 26.4 weeks of gestation. Thereafter the mean size of the largest cyst within the CCAM was decreased.
We also assessed the fetal left cardiac function in cases 5 and 6, and the Mod-MPI before and after these procedures is indicated in Table 2. Both fetuses demonstrated improved cardiac function after these intrauterine treatments.
Table 3 demonstrates the postnatal outcomes of the six fetuses with macrocystic CCAM. The median gestational age at delivery was 38.0 weeks (range, 32.4-40.3 weeks), and the mean birth weight was 3121±543 g. The male to female ratio was 5:1. Four of these newborns showed respiratory symptoms at birth, and three infants required chest tube insertion because of these developing symptoms. All six babies underwent the surgical resection at a median age of 6 days (range, 1-136 days) and were confirmed with macrocystic CCAM. Based on the postnatal follow up in each case, these children are currently all doing well without any complications.
Discussion
Our study demonstrated that intrauterine therapy, especially thoracoamniotic shunting can save the fetuses with macrocystic CCAM even who developed hydrops. On the contrary to microcystic CCAM, which tend to shrink [4,10], macrocystic lesions may grow or show few changes during the third trimester [9], and may also cause a cardiac compression with changing hemodynamics and hydrops because of an elevated central venous pressure [11]. Previous reports have often described a high incidence of adverse features such as fetal death or hydrops in macrocystic CCAM [4]. Therefore, these lesions may require intrauterine therapy including aspiration of the cyst, thoracoamniotic shunt, injection of sclerosing agent, or in some cases open fetal surgery to prevent such complications [12]. In our experience, the cyst was reduced or completely removed after aspiration and thoracoamniotic shunting. Fetal hydrops and fetal cardiac functions were also improved after successful intrauterine treatments.
CCAM is generally classifies into three major subtypes: type I, macrocystic, contain one or more large cysts >2 cm and a compressed normal parenchyma; type II, several cysts, of <1 cm in size and blending with the adjacent normal parenchyma; and type III, microcystic consisting of several noncystic masses [13,14]. More recently, two additional subtypes of CCAM have been added to the classification of this disorder, in accordance with the origin site of the malformation: These are type 0 which involves acinar dysplasia and type IV which denotes a peripheral cyst of the distal acinus with alveolar type cells [15]. Because this original CCAM classification is based on histology, it cannot be applied prenatally [13]. To overcome this limitation, Adzick et al. [16] proposed a new classification system for CCAM based on the predominant component of the lesion (cystic or solid). These authors classified the disease as either macro- or microcystic in appearance based on gross anatomical observations and ultrasound. A macrocystic lesion denotes single or multiple cysts that are 5mm or larger in diameter whereas a microcytic lesion is a solid echogenic mass. This new approach by Adzick et al. [16] has become the accepted classification system date.
There are several prognostic markers of the postnatal outcomes of fetuses with CCAM. These include the size and type of CCAM, the presence of a mediastinal shifting, polyhydramnios, or hydrops and CCAM volume ratio (CVR) [6,11,12]. CVR is defined as the CCAM volume (mL) to head circumference (cm) ratio. The fetus with CVR greater than 1.6 is increased risk of hydrops, whereas the fetus with CVR less than or equal to 1.6 is presented to be low risk of hydrops [6]. Therefore, fetuses with these poor prognostic markers should be considered for intrauterine therapy.
However it must be noted that there are currently no universal guidelines for fetal therapy of a large cystic lung lesion. Fetal interventions in such cases can be based on gestational age, the size of the CCAM, and the presence of poor prognostic markers. If the cyst fluid reaccumulates rapidly in a fetus of under 32 weeks gestation, fetal thoracoamniotic shunting could be considered as an effective and appropriate intervention [16]. Since Clark et al. reported the first successful shunting operation for macrocystic CCAM [11], several cases successfully treated in this way have been reported from other countries [9]. We have also reported two cases of successful in utero fetal shunting to treat macrocystic CCAM, the first such report from Korea [17]. Our current study reports four additional cases of macrocystic CCAM since then, and showed the favorable postnatal outcomes.
In cases of macrocystic CCAM for which an enforced shunting operation is not feasible, percutaneous fetal sclerotherapy are possible alternatives. Intrauterine sclerotherapy has been used to treat fetal pleural effusion, cystic hygroma, and bronchopulmonary sequestration [18,19,20]. Recent reports also demonstrated that complicated CCAM can be managed by fetal sclerotherapy and they concluded this treatment modality is minimally invasive and highly effective [21,22]. OK-432, which is widely used as a safe and effective sclerosing agent, can break down the normal epithelia of cystic walls via inflammatory reactions and the resulting fibrotic adhesions cause lymphatic fluid reduction and shrinkage [23]. KE is used as a unit of measurement for OK-432 doses and 1 KE corresponds to 0.1 mg of freeze-dried streptococci containing approximately 1×108 cells [24]. Although the recommended dosage for OK-432 is 0.1 KE per injection [18], we started from the minimal dose and repeated with higher dose because of reaccumulation of fluid within the cyst. Our use of OK-432 was based on the positive results of previous studies, but we did not observe the optimal outcomes. However, the ineffectiveness of OK-432 cannot be based on only one case, and further assessments of this treatment in additional CCAM cases will be needed to draw firmer conclusions.
A limitation of our study is the small cohort of only six fetal cases. However, the incidence of CCAM is already rare, and the proportion of these cases with macrocystic CCAM is very small. In addition, although CVR is considered as a useful prognostic indicator of fetal hydrops, we measured only two dimensions of the CCAM in this study. Measuring the cross-section (width×length) of the CCAM instead of its volume make it difficult to evaluate the effectiveness of a given intervention. Therefore there is a future need for measurement standards to be developed. Despite of these limitations, our current results suggest that intrauterine therapy for macrocystic CCAM produces a good outcome. In addition, our present findings may provide useful information to assist the future design of sclerotherapy interventions for macrocystic CCAM.
We concluded that prenatal intervention is effective treatment in macrocystic CCAM fetuses who have poor prognostic markers. Therefore we suggest these intrauterine decompression therapies as recommendable interventions for macrocystic CCAM fetuses.










")











