


Introduction
Prenatal testing is an integral component of obstetric practice, and is commonly performed in professional medical organizations. The primary aim of prenatal testing is the diagnosis of fetal aneuploidies, such as trisomy 21 (T21, Down syndrome), trisomy 18 (Edwards syndrome), and trisomy 13 (Patau syndrome), as well as aneuploidies related to the X and Y chromosomes [1]. Although the majority of fetuses with aneuploidy result in termination during the development of the fetus, T21 has the highest survival rate, which affects 1 in 800 births [2]. Therefore, the prenatal detection of T21 is considered the most common and important aspect of prenatal genetic testing.
Prenatal testing of T21 falls into 'screening' and 'diagnosis' category. Current prenatal screening tests have greatly improved by using a combination of maternal serum markers and fetal sonographic markers such as nuchal translucency [3-6]. The best performing screening tests are able to identify more than 90% of T21 cases, with a 5% rate of false positives. However, positive screening results require confirmation with diagnostic testing, such as amniocentesis or chorionic villus sampling (CVS). The accuracy of these diagnostic methods is estimated to be 98% to 99% [7]. However, both sampling procedures are invasive, and are associated with significant risks to the fetus and mother, including the potential loss of a healthy fetus [7,8]. For this reason invasive prenatal diagnosis tests are currently preformed only in high-risk pregnancies or in pregnancies with increased maternal age and/or family history of having a child with an inherited disease. Therefore, developing a reliable method for non-invasive prenatal diagnosis (NIPD) for fetal T21 is of critical importance in prenatal care.
To perform NIPD, a source of fetal genetic material that could be sampled without harm to the fetus would be needed. Since the 1970s, researchers have isolated intact fetal cells in maternal circulation [9]. However, fetal cells in maternal blood are rare in quantity and tend to remain in the mother's body for years [10]. Hence, this method is unsuitable for NIPD [11]. In 1997, Lo et al. [12] discovered the existence of cell-free fetal DNA (cff-DNA) in maternal circulation. Compared to fetal cells, cff-DNA is relatively more abundant in maternal blood and thus has been regarded as a promising new material for NIPD. It constitutes approximately 10% of the total DNA in maternal plasma and is rapidly cleared from maternal blood, within two hours of delivery [13,14]. Moreover, it has recently been found that the entire fetal genome, in the form of cff-DNA, is present in maternal blood [15]. Therefore, cff-DNA has become the focus of research for the development of NIPD.
Currently, the clinical potential of cff-DNA has been demonstrated. In particular, the determination of fetal sex and fetal Rhesus D status using cff-DNA is already applied as routine tests in Denmark, Sweden, and the Netherlands [16-18]. However, the application of cff-DNA for the NIPD of fetal T21 has been considered to be technically challenging. One aspect of the challenge relates to the presence of the large amount of maternal DNA which interferes with the analysis of the fetal DNA in maternal plasma [13]. Another challenge is related to the characteristics of the cff-DNA that pose a difficulty in determining the chromosome dosage of the fetus. Recently, various methods have been applied to overcome these challenges in the NIPD of fetal T21 using cff-DNA and promising results have been reported. In this review, we discuss the most recent technologies for the NIPD of fetal T21 using cff-DNA, and their use in clinical practice.
NIPD of Fetal T21 Using Single-molecule Counting Methods of Chromosome Sequences
The need for reliable methods for NIPD of T21 has created a strong interest in the field of rapid and accurate single-molecule counting methods (e.g., digital polymerase chain reaction [PCR] and massively parallel DNA sequencing [MPS]), which could be used in routine clinical diagnosis in the form of automated platforms [19]. The single molecule counting techniques can detect fetal aneuploidy without the analysis of fetal-specific DNA in maternal plasma [20]. These methods are based on the detection of the extra copy of chromosome 21 to distinguish normal cases from T21 cases. For example, in cases where a woman is carrying a fetus with T21, the number of copies of chromosome 21 in the maternal blood is expected to be slightly higher in comparison with other autosomes. Currently, rapidly developing MPS technologies, which provide a vast amount of data across the entire genome, appear to be suitable for counting genome representation and determining the over-represented chromosomes 21 in the affected fetus. Moreover, these techniques can simultaneously detect all trisomies in a single step.
Digital PCR
Digital PCR is a single molecule counting technique that allows the quantification of DNA by counting one molecule at a time. It has superior analytical precision compared with conventional PCR methods. Thus, it can precisely quantify small increments within the total (maternal+fetal) amount of DNA molecules derived from chromosome 21 for T21, when compared with euploid pregnancies. Lo et al. [21] reported on the application of digital PCR for the NIPD of T21. They used an approach called relative chromosome dosage where the amount of plasma DNA molecules from chromosome 21 was compared with that of a reference chromosome, that is, a chromosome expected to have a normal dosage in the fetus [21]. The relative chromosome dosage of chromosome 21 to the reference chromosome was elevated in maternal plasma of women with T21 fetus and the degree of increments was dependent on the fetal DNA concentration. However, in the application of digital PCR for the NIPD of fetal T21, the analytical platform of digital PCR needs to be quantitatively more precise to reliably determine the small expected increment. Quantitative precision can be improved by increasing the number of PCR analyses performed. A previous study has shown that the accurate detection of fetal T21 in a maternal plasma sample containing 25% fetal DNA requires approximately 8,000 digital PCRs [21]. Therefore, the clinical setting for the NIPD of fetal T21 using digital PCR may require the use of automated platforms.
Next-generation DNA Sequencing
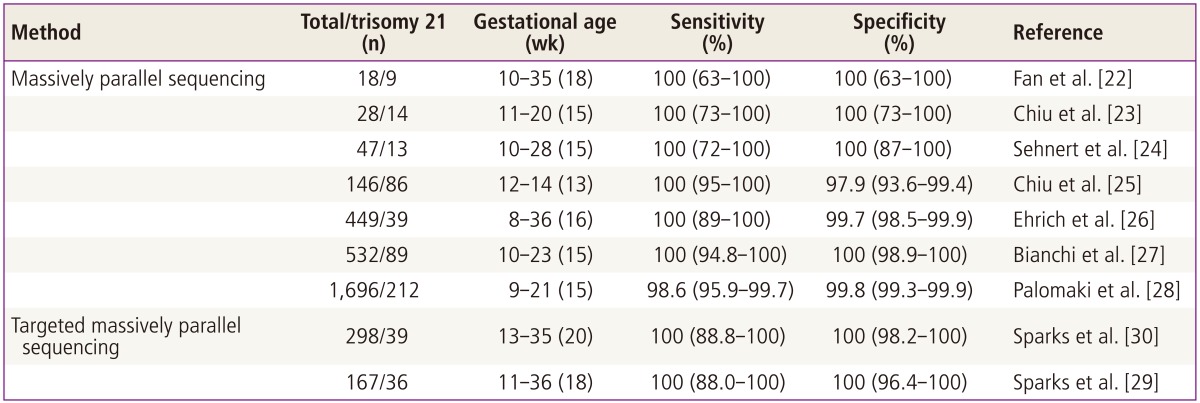
New next-generation DNA sequencing (NGS) technologies permit the simultaneous sequencing of extremely large quantities of DNA molecules. NGS produces millions or billions of short sequence reads per instrument run. NGS of cff-DNA from maternal blood has enormous potential, not only for increasing our understanding of the causes of prenatal genetic disorders in the fetus but also for designing non-invasive clinical diagnostic tests [15]. The possibility of using NGS to detect non-invasive fetal trisomy from maternal blood has been demonstrated [22-24], and this finding has been confirmed in other recent studies (Table 1) [25-30]. An alternative approach to sequencing whole genomes for the non-invasive detection of fetal abnormalities is to enrich only interest regions prior to sequencing [29-31]. Moreover, NGS technologies show remarkable potential for detecting the most common aneuploidies, including T21, T18, and T13. Currently, these discoveries have been translated into clinical tests, resulting in major benefits for NIPD.
Generally, the NIPD of fetal T21 using NGS is done through the following process. First, a short region at one end of each DNA molecule of maternal plasma is sequenced using synthesis technology and mapped against the reference human genome to determine the chromosomal origin of each sequence. Next, the density of the sequenced tags from the chromosome 21 of interest from a T21 fetus is compared with cases of trisomy and euploid pregnancies. Consequently, NGS can clearly identify samples from women carrying aneuploid fetuses by comparing them with samples taken from women with known euploid fetuses. Previous studies demonstrated that NGS was highly accurate in the direct detection of fetal T21 from maternal plasma (Table 1) [22-30]. The accuracy of NGS for the NIPD of T21 has already been validated by large-scale clinical studies. However, sequence information of NGS is obtained for the various chromosomes proportional to their sizes.
Therefore, chromosome 21, being the smallest autosome, would only be represented by a relatively small percentage of the sequence reads. As a result, the throughput of NGS for NIPD of fetal T21 is too low. To overcome the limitations of NGS, several targeted sequencing approaches were developed based on the a priori selection of DNA regions for analysis. Compared to sequencing and counting all reads from chromosomes, limiting the number of DNA regions greatly reduces the effort required to assess the dosage of a chromosome. Moreover, the careful selection of the DNA regions to quantify can potentially reduce the confounding variation in the number of reads per locus by taking into account only the loci with similar properties (e.g., GC content or the number of repeats of a particular sequence in the genome) [29,30]. Sparks et al. [30] described a method for detecting chromosome aneuploidy using NGS combined with an amplificationbased enrichment assay. The assay is comprised of three oligos per analyzed locus. Of the 298 samples, including 39 trisomy 21 samples and seven trisomy 18 samples, all aneuploidy samples were correctly distinguished from the controls, and as such the authors concluded the assay to have 100% sensitivity and specificity. The level of sequencing, covering only 420,000 reads per sample, was nevertheless sufficient to detect trisomy 21 and trisomy 18 reliably (z statistics exceeded 3.6 in all samples). This level corresponds to a <5% of the level required by non-targeted approaches. Moreover, this method enables multiplexing (96 samples were processed simultaneously), thus greatly reducing the cost of the analysis. The recent single nucleotide polymorphism (SNP)-based targeted NGS method was developed for the NIPD of fetal aneuploidies [29,30] and seems to be highly efficient. The key feature of this method is that it takes the mixture of maternal and fetal DNA obtained from blood plasma into account, separately from the DNA from one or both parents. Along with T21, T18, T13, and sex chromosome aneuploidies (e.g., X0, XXY, XXX, XYY) can also be detected, which is an important advantage of this method in light of the high occurrence of these abnormalities. A clinical trial of the prenatal non-invasive aneuploidy testing using SNPs, supported by the National Institutes of Health, is currently underway [32]. As it is SNP-based, the method may need to be tested on patients from different populations. Nevertheless, as targeted DNA sequencing can be performed on a sequencing machine with a lower price per run and lower throughput (e.g., PGM, Ion Torrent [Life Technologies, San Francisco, CA, USA], or MiSeq [Illumina Inc., San Diego, CA, USA]), these methods are preferred, especially for average-sized clinics.
Epigenetic Approaches for the NIPD of Fetal T21
The major challenge for the development of NIPD using cff-DNA is that cff-DNA only constitutes around 10% of the total DNA in the maternal circulation [13]. To differentiate the fetal-derived sequences from that of the mother, the most intuitive targets for the detection of fetal DNA were based on absolute discriminative genetic markers, such as Y-chromosome-specific loci or paternally-inherited polymorphic loci that are either absent or different in the maternal genome [33-35]. However, these types of fetal markers were associated with certain limitations in practice. Firstly, diagnostic tests developed based on Y-specific targets could only be applied to pregnancies involving male fetuses. Secondly, the detection of a paternally inherited polymorphism requires prior knowledge of the polymorphic status of the parents, and could only apply to a subset of individuals who possessed that particular polymorphism. Therefore, it would be desirable to develop a type of marker that allows for a confident differentiation of the fetus from the mother, and yet is independent of the gender or polymorphic status of the fetuses. Recently, epigenetic modifications as fetal-specific signatures to detect cff-DNA from circulating maternal DNA have been investigated.
Fetal-specific Epigenetic Makers for NIPD
Epigenetic modifications refer to inheritable molecular processes that affect gene expression without changing the DNA sequence or content, and the most widely studied epigenetic process is DNA methylation. The possibility of DNA methylation as a non-invasive biomarker was first demonstrated in the plasma of patients with cancer [36-38]. Soon after such discoveries, various attempts have been made to identify fetal-specific epigenetic markers based on differential methylation patterns between the fetus and the mother [39-41]. Fetal-specific methylation pattern is divided to parent origin-specific methylation pattern and placenta specific methylation pattern.
First, parent origin-specific methylation pattern is based on genomic imprinting in humans [42,43]. Fetal epigenetic markers are developed with an imprinted region, in which the DNA methylation patterns are inherited in a parent origin-specific manner [44]. For example, if a pregnant woman has inherited the methylated copy of an imprinted region from her father, an imprinted region in her fetus would become unmethylated because she passed. The methylation status of this region is distinguishable between the fetus and the mother in an allele-specific manner. In 2002, the imprinted region between the IGF2 and H19 genes was investigated to detect fetal-specific methylation from maternal plasma [39] and was confirmed by genotyping a biallelic polymorphism within the differentially methylated regions [39]. However, this method would be relatively complicated to use as a routine fetal marker, because this marker was based on an imprinted locus. Next, placenta specific methylation pattern is based on the human placenta with a specific DNA methylation pattern that is different with somatic tissues [45-47]. The majority of cff-DNA in the maternal plasma was derived from the placenta, while the maternal cell free DNA in the maternal plasma was predominantly derived from the maternal hematopoietic cells [48-50]. Therefore, genomic regions that are differentially methylated between the placenta and the maternal blood cells have been considered as fetal-specific epigenetic makers in maternal plasma. In 2005, a region on the maspin (SERPINB5) gene promoter was firstly found to be hypomethylated in the placenta, while hypermethylated in the maternal blood cells [40], and the hypomethylated sequences of the SERPINB5 gene were detectable in maternal plasma throughout the course of pregnancy, and its level dropped significantly after delivery. Therefore, this was reported as the first universal fetal marker that can be used in all pregnancies, regardless of fetal gender and genotype. After this discovery, various attempts were made to identify a number of genomic regions that are differentially methylated between the placental tissue and the maternal peripheral blood cells according to the principle of NIPD. This feature allows for the development of a single, simple test to determine the presence of cff-DNA in the maternal plasma with greater simplicity and coverage. The approaches used for the detection of these markers are variable, depending on whether the placental-derived sequences are hypermethylated or hypomethylated compared with the maternal blood cells.
Detection Method of Fetal Epigenetic Markers
To detect fetal epigenetic markers in maternal plasma, the first step is to differentiate methylated and unmethylated sequences. Various methods, such as a bisulfite modification of the template DNA, differential cleavage by restriction enzymes and antibody-mediated enrichment of methylated fragments by methylated DNA immunoprecipitation (MeDIP), are applied. The next step is to quantify a fetal-specific methylation pattern. In general, PCR-based methods, such as quantitative methylation-specific PCR and quantitative real-time PCR, are used.
Briefly, the process of bisulfite conversion changes unmethylated cytosine residues into uracil, leaving methylated cytosine unchanged [51]. The bisulfite-converted DNA is differentially amplified by PCR-based methods, depending on the methylation status of the regions where the primers bind [52]. However, bisulfite DNA conversion results in the degradation of >90% of the template DNA [53]. Therefore, this technique is undesirable for the detection of cff-DNA, which is present at a lower abundance in maternal plasma, particularly during early gestation. Methylation sensitive restriction enzymes, such as BstU I or Hpa II, can also be distinguished to differentiate between methylation patterns in DNA sequences. These restriction enzymes sensitively digest ummethylated cytosine bases in their recognition sequence, such as CGCG or CCGG. To quantify cff-DNA in maternal blood using methylation-sensitive restriction enzymes, cell-free maternal DNA should be unmethylated. This unmethylated maternal DNA is removed in cell-free total plasma DNA by the treatment of such enzymes, and then can be quantified the digestion-resistant (methylated) cff-DNA by quantitative methods, including real-time PCR or digital PCR [41,54]. Compared with bisulfite conversion, this digestion-based method introduces less damage to the plasma DNA. However, the enzyme cleavage effectiveness, depending on the duration of digestion or the amount of enzymes used, can affect the quantification of cff-DNA [55]. Recently, MeDIP, which captures DNA containing methylcytosine, has been applied to quantify cff-DNA. This method can capture only methylated DNA fragments using a monoclonal antibody specific for methylcytosine and provides up to a 90-fold enrichment of methylated DNA. Generally, the unmethylated or methylated DNA sequences can be quantitatively measured by a methylation-specific PCR (MSP) using a fluorescence probe. The copy number is calculated directly from the amplification curves of the fluorescence signal by a series of calibration standards. This method has been widely used to identify methylation patterns of cff-DNA in maternal plasma [56,57] and applied to develop effective epigenetic tests for the NIPD of fetal T21.
Potential of Fetal-specific Epigenetic Marker in NIPD of Fetal T21
Analysis of differences in the DNA methylation patterns between the maternal and fetal circulating DNA molecules has been proposed as an alternative strategy to the analysis of cff-DNA sequences in the NIPD of fetal T21. Such epigenetic markers could be useful either via the analysis of the epigenetic allelic ratios or directly compared with a placenta-derived DNA methylation marker on a reference chromosome.
The fetal-specific epigenetic markers require:1) the detection of a number of DNA sequences that are differentially methylated between maternal and fetal DNA and 2) quantification of these fetal-specific DNA sequences by methods such as quantitative MSP or quantitative real-time PCR. Previous studies described that PDE9A on 21q22.3, which were hypomethylated in the placental tissues while completely methylated in the maternal peripheral blood cells, can be used for the NIPD of T21 [58,59]. The putative promoter regions of HLCS on 21q22.13, which are hypermethylated in the placental tissue compared with the maternal blood cells, are also applied for the NIPD of fetal T21 and have reported promising results [60]. Theoretically, the allelic ratio of a fetal-specific epigenetic marker may present equal signal intensity for unaffected fetuses and an increased signal intensity of chromosome 21 for T21 fetuses. Using this approach, fetal T21 can be detected non-invasive even during the first trimester [42,60]. The enrichment of sequences which are specifically methylated in the placenta and/or the analysis of multiple informative markers on the chromosome 21 have been applied to detect fetal T21 with high sensitivity and specificity. Recently, various methylation-specific techniques, such as antibody-mediated enrichment of methylated fragments by MeDIP and differential amplification of methylated fragments via Hpa II tiny fragment enrichment by ligation-mediated PCR (HELP), were used for the NIPD of fetal T21 using fetal epigenetic markers [61-64]. The correct diagnosis in the NIPD for fetal T21 using fetal epigenetic markers is based on the ratio of a subset of fetal-specific methylated regions located on chromosome 21 compared with normal cases. This new platform is calculated with further statistical analysis of multiple markers and has exhibited excellent clinical performance (both the sensitivity and specificity were 100%) [65]. This methodology seems to be easily reproducible and can be readily performed by equipment currently present in most diagnostic laboratories without sophisticated analytical platforms. Moreover, this approach can be simultaneously detected in all known aneuploidies, if regions exist where the fetal DNA is hypermethylated compared to the maternal peripheral blood DNA are provided. Therefore, this technique seems to have the right properties to become a NIPD technique for T21 and would provide a cost-effective alternative. However, such an approach is limited in the practical applicability of NIPD for fetal T21 because of the low number of copies of cff-DNA in maternal blood and the variability in the levels of DNA methylation of individual fetal-derived epigenetic markers can affect the results and its clinical value remains to be proven in large-scale clinical studies.
Conclusion
The development of an NIPD technique for fetal T21 that would provide true genetic information without carrying risk for the progress of the pregnancy will continue to be an actively researched area in prenatal diagnosis. Trials performed so far highlight the medical and commercial potential of NIPD, but the proposed techniques warrant further validation in clinical practice. Throughout the last decade, considerable achievement has been made regarding the technical possibilities for the NIPD of T21. In the previous years, male-specific signals or paternally inherited polymorphisms have been proposed as targeted fetal DNA markers, but research interest has now evolved to the detection of fetal-specific patterns or epigenetic signatures with a unique methylation pattern that will allow the application of NIPD in all pregnancies. In parallel, novel sequencing methods with high diagnostic accuracy have already been applied in the clinical setting as an effective breakthrough for the NIPD using cff-DNA. Yet, population-based, double-blind, large-scale clinical trials are required to verify the diagnostic potential of these methods and their cost-effectiveness compared with the conventional screening tests before their introduction into the clinical practice of fetal medicine. In particular, the fact that NIPD using cff-DNA requires a small sample of maternal blood may create numerous ethical, social and legal implications, owing to the ease with which the test can be performed. Therefore, the use of this method should be carefully considered in clinical situations. Nevertheless, in the near future, the NIPD of fetal T21 using cff-DNA will be applied in the clinical setting as an effective choice for all pregnant women who opt for safer prenatal diagnostic testing. Eventually, the availability of a reliable non-invasive test to determine fetal T21 would reduce unintended fetal losses and would presumably be welcomed by pregnant women.






")











