Differential expression and methylation of integrin subunit alpha 11 and thrombospondin in the amnion of preterm birth
Article information

Abstract
Objective
This study aimed to investigate the association between preterm birth and epigenetic mechanisms in the amnion.
Methods
We examined the association between differentially methylated regions (DMRs) and differentially expressed genes (DEG) using a cytosine-phosphate-guanine methylation array and whole-transcriptome sequencing from the amnion (preterm birth, n=5; full term, n=5). We enrolled 35 participants for mRNA expression analysis and pyrosequencing: 16 full-term and 19 preterm subjects. We compared the association of integrin subunit alpha 11 (ITGA11) and thrombospondin 2 (THBS2) gene methylation status with mRNA expression in the amnion.
Results
In the preterm birth group, methylation of ITGA11 and THBS2 genes was significantly lower (ITGA11 gene: 60.30% vs. 73.16%, P<0.05; THBS2 gene: 64.59% vs. 73.16%, P<0.05), and the expression of the genes was significantly higher than that in the full-term group (ITGA11 gene: 14.20 vs. 1.57, P<0.01; THBS2 gene: 1.18 vs. 10.34, P<0.05).
Conclusion
Methylation of the ITGA11 and THBS2 genes in the amnion was associated with preterm birth. Thus, ITGA11 and THBS2 gene methylation status in the amnion may be valuable in explaining the mechanism underlying preterm birth.
Introduction
Preterm birth, defined as delivery before 37 weeks of gestation, is associated with higher neonatal death rate [12]. Preterm birth rate increased from 24.15% in 1995 to 53.70% in 2010 [3]. Preterm babies show increased risk of mortality in the first year of life, neurodevelopmental disabilities, and behavioral problems [45], as well as development of hypertension, type 2 diabetes, and cardiovascular disease in the future [2].
The fetal membranes consist of two distinct layers (the amnion and the chorion). They are developed from extra-embryonic tissues consisting of fetal (amniotic fluid) and maternal components (maternal decidua) [67]. The amniotic membrane (AM), or amnion, is a thin membrane on the inner side of the fetal placenta [8]. The AM provides most of the tensile strength in the fetal membranes with an important role in the maintenance of pregnancy and parturition [9]. During pregnancy, the AM is important because it reduces scarring and inflammation [7]. The premature rupture of fetal membranes (PROM) is an event integral to the onset and development of preterm labour (PTL) [10]. Generally, the physiology of ROM in preterm birth has been attributed to cellular apoptosis, extracellular matrix (ECM) remodeling, and stretch-induced physical weakening of the fetal membranes [1011].
Recently, a new hypothesis has suggested that multiple factors can increase the risk associated with preterm birth through independent biological mechanisms, such as genetic factors and epigenetic effects (e.g., DNA methylation) [12]. Generally, DNA methylation is an important epigenetic event regulating chromatin structure and gene expression that can induce changes in the phenotype of a cell or an organism without altering its DNA sequence [13]. Some researchers have investigated the informative locations for DNA methylation changes at promoters, cytosine-phosphate-guanine (CpG) islands, and neighboring regions, and at loci, such as imprinted differentially methylated regions (DMRs) using microarray methods [14].
Some studies have shown significant differences in methylation values associated with gestational age and preterm birth [215161718] using the myometrium [19], amnion [9], and blood (maternal and cord) samples [20]. Recently, epigenetics has become an active area of investigation, and the prevalence of preterm birth has increased [21]. However, the association between epigenetic mechanisms in the amnion and preterm birth remained unclear. We hypothesized that DNA methylation is associated with the weakness of amnion cells, leading to preterm premature rupture of membranes (PPROM) or PTL. Thus, we examined the association of DMRs with differentially expressed genes (DEG) between the amnions of women with term and preterm birth using a DNA methylation array, whole-transcriptome sequencing (WTS), pyrosequencing, and gene expression data.
Materials and methods
1. Study subjects
This study was conducted as part of the Ewha Preterm Birth and Case–Control Study, a birth cohort established at the Ewha Womans University Hospital, Seoul, Republic of Korea, between June and December 2014. The study was approved by the Institutional Review Board of the Ewha Womans University Hospital (ECT 06-127-7). All participants provided informed consent, and the study was approved by the hospital's Institutional Review Board. Amnion samples from singleton pregnancies with PTL and/or PPROM were collected during delivery, and we followed the birth outcome. Women with multiple births, major birth defects, and pregnancy complications were excluded. Gestational age was determined using the first day of the last menstrual period and ultrasound results.
2. DNA preparation and DNA methylation array
Amnion samples were collected from pregnant women with term and preterm pregnancies at the Ewha Womans University MokDong Hospital. Genomic DNA was isolated from the amnion samples of term (n=5) and preterm (n=5) deliveries using the QIAamp DNA mini kit (Qiagen, Hilden, Germany), quantitated with a NanoDrop device, and checked for integrity with 1% agarose gels. For each sample, up to 700 ng of genomic DNA was bisulfite-converted using the Zymo EZ DNA methylation kit (Zymo Research, Orange, CA, USA), amplified, fragmented, and hybridized on an Illumina Human Methylation 450 BeadChip (Illumina, San Diego, CA, USA) according to the manufacturer's protocol. Briefly, bisulfite-converted DNA was fragmented with the fragmentation mix buffer supplied with the kit and hybridized to beads on chips that contained appropriate probe sequences. After hybridization, single-nucleotide extension was performed, with the DNA fragments hybridized on the BeadChips, which were washed before scanning.
After washing, the BeadChips were scanned with a HiScan SQ system (Illumina). Scanned images were processed, and signal intensity was calculated using the GenomeStudio software (Illumina). The 450K Infinium Human Methylation 450 BeadChip microarray was processed with the GenomeStudio Methylation Module, which was used to quantify the methylation level, in a range from 0 to 1. DMRs were determined through the Diff Score (transformation of the P-value).
3. Whole-transcriptome sequencing analysis
Total RNA was isolated from the amnion samples of women with term (n=5) and preterm (n=5) deliveries using the TRIzol reagent (Life Technologies, Carlsbad, CA, USA). RNA integrity was confirmed with a Bioanalyzer (Agilent, Santa Clara, CA, USA) using the RNA 6000 Nano Kit (Agilent). Isolated total RNA was processed for preparing the RNA sequencing library using the TruSeq stranded total RNA sample preparation kit (Illumina) according to the manufacturer's protocol. Briefly, rRNA samples were depleted from 1 μg total RNA using rRNA removal beads, followed by enzyme shearing. After first- and second-strand cDNA syntheses, A-tailing and end repair were performed for the ligation of proprietary primers that incorporate unique sequencing adaptors with indexes for tracking Illumina reads from multiplexed samples run on a single sequencing lane. For each library, an insert size of up to 200 bp was confirmed with a Bioanalyzer using a DNA kit (Agilent), and the library was quantified using real-time polymerase chain reaction (PCR) with a CFX96 real-time system (BioRad, Hercules, CA, USA). Sequencing of each library was performed on an Illumina NextSeq500, and clusters of the cDNA libraries were generated on a TruSeq flow cell and sequenced for 100-bp paired end reads (2×100) with a TruSeq 200 cycle SBS kit (Illumina). The raw image data were transformed by base-calling into sequence data and stored in FASTQ format. The WTS reads were mapped onto the human (hg19) reference genome and assembled from alignments for transcript structures using the Tuxedo package. After assembly, differential expression analysis was performed using the Cuffdiff module of the Cufflinks package.
4. Database for Annotation, Visualization, and Integrated Discovery analysis
DEGs and DMRs were identified through the WTS and 450K microarray results. Candidates were selected when an inverse correlated relationship was satisfied with expression and methylation levels in the promoter region. To determine statistically significantly overrepresented gene ontology (GO) terms, the Database for Annotation, Visualization and Integrated Discovery (DAVID; http://david.abcc.ncifcrf.gov, ver. 6.7) was used [22], and functional annotation charts were created with default settings.
5. Pyrosequencing
CpG site-targeted bisulfite pyrosequencing and real-time PCR were used to confirm the BeadChip results in the amnion. Briefly, 20 ng of genomic DNA from each of the amnion samples from term (n=16) and preterm (n=19) deliveries was treated with sodium bisulfite using the EZ DNA methylation kit (Zymo Research) according to the manufacturer's protocol. The integrin subunit alpha 11 (ITGA11), and thrombospondin 2 (THBS2) genes were amplified using a forward primer and a biotinylated reverse primer designed by PSQ Assay Design (Biotage AB, Uppsala, Sweden) and amplified using a GeneAmp PCR system 9700 (Applied Biosystems, Waltham, MA, USA). The PCR reactions were performed at an annealing temperature of 60°C for 35 cycles. Pyrosequencing reactions for ITGA11 and THBS2 gene methylation were conducted with sequencing primers on the PSQ HS 96A System (Biotage AB) according to the manufacturer's protocol (Fig. 1).
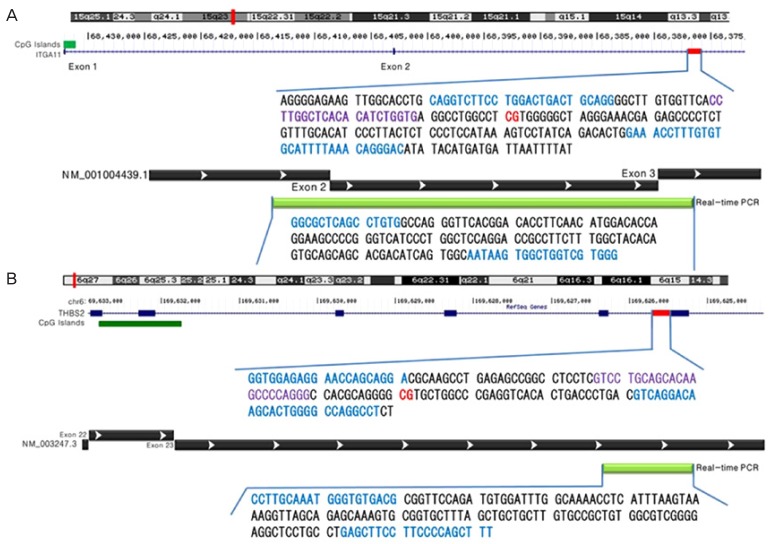
Schematic representation of the human integrin subunit alpha 11 (A) and thrombospondin 2 (B) genes. Red line bar: pyrosequencing location; blue font: forward and reverse primer regions; violet font: sequencing primer region; green bar: real-time polymerase chain reaction location.
The percentage of methylation was calculated by dividing the peak area of the methylated cytosine (mC) peak by the combined peak areas of the non-methylated cytosine (C). The methylation index of each sample was calculated as the average value of mC (mC+C) for all examined CpGs in the target region.
6. Real-time polymerase chain reaction
For quantitative real-time PCR, 1-μg RNA was converted to cDNA using SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA, USA) and RNasin (Promega, Madison, WI, USA) in a 20-μL reaction mixture. Real-time PCR was then performed in a 20-μL reaction mixture containing cDNA, 200-nM primers for each gene, SYBR Premix EX Taq (Takara Bio, Shiga, Japan), and ROX reference dye (Takara Bio) using a PRISM 7000 sequence detection system (Applied BioSystems, Foster City, CA, USA). Briefly, the samples were heated to 95°C for 10 minutes and then amplified for 40 cycles at 95°C for 15 seconds, with annealing at 62°C for 1 minutes, followed by a dissociation stage at 95°C for 15 seconds and 62°C for 20 seconds per cycle. Quantities of each gene were calculated using the ΔΔCT method and based on the cycle threshold (CT) normalized against glyceraldehyde-3-phosphate dehydrogenase. The primer sequences for pyrosequencing and real-time PCR are listed in Table 1.

List of primers used in this study
7. Statistical analysis
The basic characteristics of the study groups were compared using Student test and χ2 test for continuous and categorical variables, respectively. To screen candidate genes, we used t-tests for methylation and expression levels and Pearson correlation tests for associations between methylation values and expression differences. We adjusted parity for differential methylation and expression between the amnion of term and preterm birth using multivariate analysis of variance. After pyrosequencing, the selected DNA methylation sites and expression levels were compared between the groups using the Mann-Whitney U test. All statistical analyses were conducted using the SAS software (ver. 9.3; SAS Institute, Inc., Cary, NC, USA).
Results
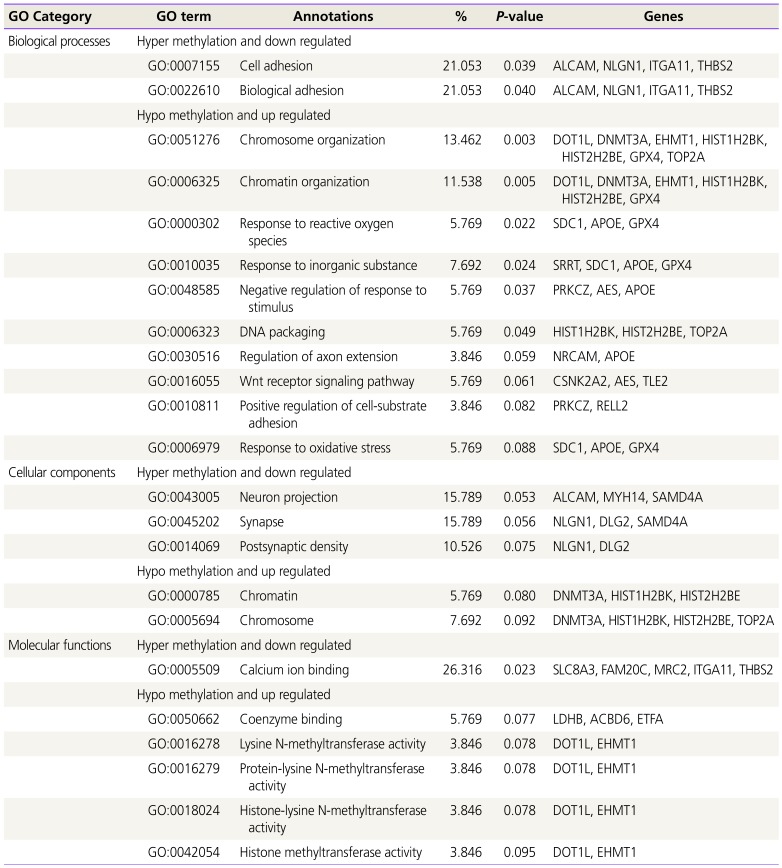
We observed significant differences in DNA methylation and RNA expression in the amnion samples between preterm and full-term birth deliveries (Fig. 2). In total, 35,850 methylated CG sites were significantly different in the amnion from term and preterm births (P<0.05, Fig. 2A). Furthermore, 1,002 genes were significantly down- or upregulated in the amnion between term and preterm births (P<0.05, Fig. 2B). Seventy-one genes were common in the methylation and expression analyses, with a negative correlation. Of the 71 differential genes, 19 (26.76%) were hypermethylated or downregulated, and 52 (73.24%) were hypomethylated or upregulated in the amnions from preterm births.
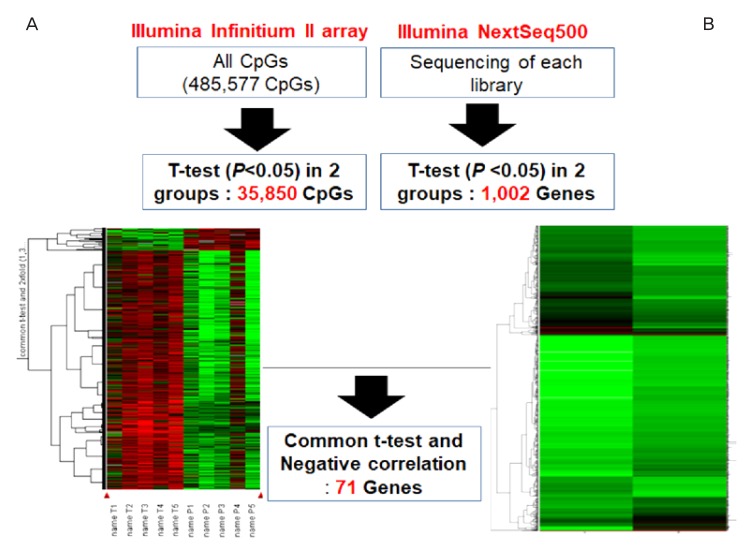
Workflow for the sequence of data analyses with array-based technology and amnion samples. (A) DNA methylation values using an Illumina Infinitium II array. (B) mRNA expression detected using next-generation sequencing technology (Illumina NextSeq 500) and the amnion samples from preterm births.
We used a GO analysis to fully understand the 71 candidate genes and their putative roles in the amnion in preterm births (Table 2). Among the GO categories, ITGA11 and THBS2 were classified as cell adhesion and biological adhesion related. This category also included the activated leukocyte cell adhesion molecule (ALCAM) and neuroligin 1 (NLGN1) genes. In the category of molecular function (MF), the solute carrier family 8-member A3 (SLC8A3), FAM20C, mannose receptor C type 2 (MRC2), ITGA11, and THBS2 genes were predicted to be related to calcium ion binding. We focused on the role of the ITGA11 and THBS2 genes related with common cell adhesion.

Functional gene ontology categories enriched in the amnion of the preterm birth group
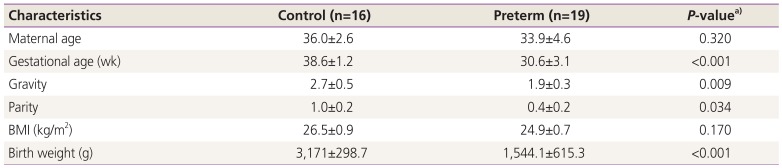
Subsequently, we validated the candidate genes by using pyrosequencing and real-time PCR. In total, 35 participants were enrolled: 16 women with term births (mean age: 36.0±2.6 years) and 19 women with preterm births (mean age: 33.9±4.6 years). Moreover, the birth weights were significantly different (term birth: 3,171±298.7 g vs. preterm birth: 1,544.1±615.3 g, P<0.001). Distributions of maternal age and BMI did not differ significantly (P>0.05) between these groups, but gravidity and parity were significantly different (all P<0.05). Detailed clinical information of the subjects is listed in Table 3.

Clinical characteristics of the validation subjects
To confirm ITGA11 and THBS2 DNA methylation values obtained from the microarray, we performed pyrosequencing in the regions, including ITGA11 (cg09430158, chr. 15: 68,375,639, GRCh38/hg38) and THBS2 (chr. 6: 169,215,780–169,254,044, GRCh38: CM000668.2). In the preterm birth group, ITGA11 gene methylation in the amnion were significantly lower (60.30% vs. 73.16%, P<0.05; Fig. 3A), and expression in the amnion was significantly higher than that in the full-term group (14.20 vs. 1.57, P<0.01; Fig. 3B). For the THBS2 gene, methylation showed significant differences between preterm and term groups (64.59% vs. 79.59%, P<0.05; Fig. 3C), with THBS2 gene expression in the amnion in the preterm group being significantly higher than that in the term birth group (10.34 vs. 1.18, P<0.05; Fig. 3D).

Methylation values of the integrin subunit alpha 11 (A) and thrombospondin 2 (B) genes in the amnion for the term and preterm births. mRNA expression levels of the integrin subunit alpha 11 (C) and thrombospondin 2 (D) genes in the amnion for the term and preterm births. The error bars represent standard deviations.
ITGA11, integrin subunit alpha 11; THBS2, thrombospondin 2.
a)P-values indicate significant differences between the term and preterm birth groups (non-parametric Wilcoxon rank sum test).
Discussion
In this study, we identified an association between the lower methylation and higher expression statuses of the ITGA11 and THBS2 genes in the amnion of preterm births using methylation array and WTS. These results suggest that ITGA11 and THBS2 gene methylation status in the amnion may be valuable in explaining the mechanism of preterm birth. We also identified at first ITGA11 and THBS2 gene methylation was associated with preterm birth in the amnion using the DMR and WTS methods.
Parturition leads to the following sequence: intact fetal membrane distention, separation of amnion from the choriodecidua with subsequent rupture of the choriodecidua, non-elastic distention of the amnion, and ultimately amnion rupture [2324]. In pregnancy, maintenance of amnion requires balance of collagen synthesis by fibroblasts [25].
In our results, the ITGA11 and THBS2 genes were associated with cell adhesion in the GO analysis. The amnion membrane contains several ECM components, such as types I, II, III, and IV collagen, laminin, and fibronectin [7]. ECM separation can decrease cell adhesion and proliferation, leading to PROM [26]. Generally, integrins and thrombospondins are known to play an important role for the regulation of cellular processes, such as cell adhesion, migration, and differentiation [2728].
The ITGA11 gene has been localized in bands q22.3–q23 on chromosome 15, and the gene encodes a mature protein with a large 1120-residue extracellular domain that contains an I-domain of 207 residues and is linked by a transmembrane domain to a short cytoplasmic domain of 24 amino acids [29]. ITGA11 has a strong affinity for type I collagen, which shows an increased expression during pregnancy and could inhibit significant tissue remodeling in the cervix [30]. ITGA11 gene expression is also increased through mid-gestation and decreased through the late stages of pregnancy [30]. The ITGA11 gene is involved in the differentiation and apoptosis pathways, leading to fibrosis in the human myometrium [31]. In addition, some integrins are activated, leading to strong adherence to the vessel wall by crosslinking via fibrinogen and fibronectin [32].
The THBS2 gene is located at the distal long arm of chromosome 6, at 6q27, and is transcribed in fibroblasts, smooth muscle cells, and osteosarcoma cell line [10]. It is a multifunctional molecule primarily described as a non-structural regulator component of the ECM, where it modulates activity and bioavailability of proteases and growth factors [33]. The THBS2 protein had a potent influence on cell adhesion, interfering with fibronectin-mediated ECM interactions [28]. These roles of THBS2 retain the complex interaction with cell surface receptors, proteases, and cytokines that characterize the mode of action of matricellular proteins [28]. Thus, ITGA11 and THBS2 gene expression may positively affect tissue remodeling or transcription events in the amnion.
We found that the ITGA11 and THBS2 genes were associated with both hypomethylation and upregulated expression in the preterm birth amnion. ITGA11 plays a multifunctional role in the recognition and organization of interstitial collagen matrices during development [34]. In addition, ITGA11 expression increased through mid-gestation, peaking on day 18 and decreasing until day 22 in the cervical tissue of rats [30]. In mice, THBS2 mRNA expression was decreased until non-pregnant levels by day 8 of gestation and remained with low level until 2 hours postpartum, returning to non-pregnant level by 24 hours of the postpartum period [35]. This conflict maybe due to the divergent role of the THBS2 in the different microenvironments according to diseases [36]. Although the expression of ITGA11 and THBS2 gene regulates cell adhesion of fibroblasts and increase of collagen, overexpression in our results proposes the increase in tissue fibrosis, resulting in preterm birth.
This study has some limitations. First, the study only included a small number of subjects: only 35 subjects were included in the preterm and term birth groups. Second, we did not assess the ITGA11 and THBS2 genes with other genes in the same pathways. Third, the ITGA11 and THBS2 genes were only confirmed on the amnion with preterm birth. Further studies using maternal blood are necessary to confirm the possibility of genetic marker.
In conclusion, methylation status and expression of ITGA11 and THBS2 genes in the amnion were associated with preterm birth. ITGA11 and THBS2 genes may involve overexpression and methylation, leading to fibrosis in the amnion. This result may be helpful in explaining the mechanism of preterm birth. Further studies are necessary to speculate an association ITGA11 and THBS2 genes with preterm birth.
Acknowledgements
This study was supported by the Ministry of Health & Welfare of the Republic of Korea (grant number: HI14C0306) through the Korea Health Industry Development Institute (KHIDI) and National Research Foundation of Korea (NRF) funded by the Ministry of Education (grant number: NRF-2016R1D1A1A09918620).
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.