



Introduction
Fetal programming occurs during embryonic and fetal development, a critical period in which tissues and organs are created. Insufficient nutrition during this time results in permanent alterations to certain structural and physiological metabolic functions of the fetus. British epidemiologist Barker first established the hypothesis, known as the "Barker hypothesis,” which states such programmed changes during this critical period predispose the fetus to certain postnatal diseases [1]. The critical period coincides with the timing of rapid cell differentiation [2]. Essentially, programming refers to the process of sustaining or affecting a stimulus or impairment that occurs at a crucial point in its development [3,4].
Rickets has long demonstrated that undernutrition in the critical early stages of life brings about a continuing change in structure [1]. A recent new doctrine suggests that fetal programming can affect diseases in adulthood. That is, the body's “memory” of undernutrition during the early stages of development translates into a pathology that determines future diseases [5]. This idea is based on animal studies that demonstrate how undernutrition in utero can alter blood pressure, cholesterol metabolism, insulin response to glucose, and other metabolic, endocrine, and immune functions important to human diseases [3,6]. This paper reviews evidence of the correlation between fetal undernutrition and diseases found in previous studies and considers the mechanism of fetal programming and the role of the placenta.
Fetal undernutrition
The nutritional status of the mother, which is an important factor that affects the programming of the body, involves factors such as maternal body composition, maternal dietary intake, blood flow to the uterus and placenta, and fetal genes. The fetus adapts to maternal malnutrition through changes in the production of fetal and placental hormones that regulate metabolism, redistribute blood flow and control growth (Fig. 1) [7]. The immediate metabolic response of the fetus to malnutrition is to consume its substrate to produce energy through catabolism [8]. Undernourishment of the fetus causes metabolic dependence on glucose to both decrease and increase the oxidation of other substrates, such as amino acids and lactic acid. Extended malnutrition results in delayed growth, reducing substrate use and lowering the metabolic rate, to improve fetal viability. Therefore, it can be hypothesized that the metabolic process of storing glucose continues into adulthood and that insulin resistance is the result of a similar process in which a decrease in the rate of oxidation in peripheral tissues that are insensitive to insulin creates increased insulin resistance. In late pregnancy, when tissues and organs are rapidly developing, any growth delays significantly affect organs and bring about disproportions in organ size. During the deceleration of growth, the fetus attempts to protect tissues, especially brain tissue, that are critical for immediate survival through redistribution of blood flow, thus resulting in a greater loss of liver and other abdominal visceral tissue [9,10]. In terms of endocrine changes, fetal insulin and insulin-like growth factor (IGF), which play a key role in growth control, are believed to respond rapidly to changes in fetal nutrition [11]. A decline in maternal food intake and the resulting drop in maternal IGF are likely to trigger decreases in fetal insulin, IGF and glucose levels. This drop reduces the transfer of amino acids and glucose from the mother to the fetus and ultimately slows the rate of fetal growth [12]. When the fetus has limited nutrient availability, anabolic growth control hormones, such as IGF-1 and insulin, decrease while the concentration levels of catabolic hormones like glucocorticoids, a major contributor to cell differentiation, increase [7,13].

Preventive epidemiological observations
A study traced 16,000 men and women born in Hertfordshire, England, from 1911 to 1930 from birth and showed that the mortality rate from coronary heart disease was twice as high in those with low birth weights than it was in the high birth weight group (Table 1) [14,15]. Birth weight and adult blood pressure also showed an inverse correlation. Prominent alterations associated with fetal programming included insulin resistance, hypertension and increased serum low-density lipoprotein (LDL) cholesterol and fibrinogen concentrations, which are all characteristics of metabolic syndrome. The odds ratio of metabolic syndrome in relation to current body mass index was 18 times higher in the lowest birth weight group than in the highest birth weight group [16].
Table 1
Death number from coronary heart disease among 15,726 men and women according to birth weight [14,15]
An association between low birth weight and coronary heart disease was confirmed in a cohort study of approximately 15,000 people born in Uppsala, Sweden [17]. The association of thin body types and type 2 diabetes was also verified with the observation that the prevalence rate of diabetes mellitus was 3 times higher in those with a low Ponderal Index (birth weight/length3) at birth. The Ponderal Index had stronger associations with diabetes than birth weight when compared to the prevalence rate of diabetes mellitus in people who had a low birth weight (Table 2) [18].
Table 2
Prevalence of type 2 diabetes by Ponderal Index at birth among 60-year-old men in Uppsala, Sweden [18]
| Ponderal Index at birth (kg/m3) | No. of men | Prevalence of diabetes (%) |
|---|---|---|
| <24.2 | 193 | 11.9 |
| 24.2 to 25.8 | 193 | 5.2 |
| 25.9 to 27.3 | 196 | 3.6 |
| 27.4 to 29.3 | 188 | 4.3 |
| ≥29.4 | 201 | 3.5 |
| All | 971 | 5.7 |
| P-value for trend | - | 0.001 |
One recent retrospective cohort study followed 13,517 patients born in Helsinki, Finland, between 1924 and 1944. This study found that not only blood pressure but also the risk of hospitalization and death from coronary heart disease were influenced by birth weights. The mortality rate from coronary heart disease was 5 times greater for those with a low Ponderal Index at birth who were overweight by age 11 when compared to those with a high Ponderal Index at birth who were thin during childhood. Their study revealed another aspect of fetal programming associated with the pace of childhood growth. Children who showed the greatest growth changes and ultimately become obese in adulthood were at the highest risk for hypertension, cardiovascular disease, and type 2 diabetes [19]. That is, those who were born small and quickly became overweight while playing “catch-up” had the greatest risk.
The results of a recent systematic analysis of 34 studies, including 66,000 people from different populations, also supported the association between low birth weight and increased blood pressure in adulthood [20]. A 1-kg change in birth weight carried a systolic blood pressure difference of 3.5 mmHg. While it does not appear significant at the individual level, this is an important change in terms of a population's mean blood pressure.
Information on the effects of fetal malnutrition during different stages of pregnancy has been gathered by studying the Dutch famine of 1944 to 1945. During the relatively short-lived Dutch famine from September 1944 to May 1945, calories consumed per day dropped to 400 to 800 kcal from the previous 1,800 kcal average then rapidly bounced back to a normal diet of more than 2,000 kcal/day post-famine. A relationship between blood pressure and low birth weight was noted but was not significant. However, further analysis revealed that adult blood pressure was associated with a significant decrease in the protein/carbohydrate ratio in the maternal diet during specific trimesters of pregnancy. In glucose tolerance tests of adults who were gestating during the famine, a blood glucose increase in relation to birth weight was observed for 2 hours. This increase was most prominent in those who had been exposed to the famine in the last trimester of pregnancy, and the effects were also more pronounced in people who were obese in adulthood. These people showed the highest head circumference/birth weight ratio and the lowest birth weights, suggesting more severe growth limitations. The least affected people were those who were exposed to the famine in the first trimester of pregnancy, but they also showed fat metabolism disorders [21].
Other adult cohorts have demonstrated signs of impaired glucose tolerance (IGT) and insulin resistance. In the Hertfordshire group, 370 men aged 59 to 70 years were tested for glucose tolerance. The prevalence of type 2 diabetes and IGT was 40% for adults with a birth weight lower than 2,500 g. This figure dropped to 14% for adults with a birth weight greater than 4,300 g (Table 3) [22]. In these associations, birth weight was independent of social class or smoking and drinking status. However, lifestyle in adulthood also could influence the effects of the intrauterine fetal environment. For example, the prevalence of IGT was highest in the low birth weight adult obesity group. Insulin resistance testing on 103 men and women in the Preston region of the United Kingdom yielded similar results, showing more insulin resistance in those with similar body weights but a lower birth weight Ponderal Index [23].
Table 3
Prevalence of type 2 diabetes and impaired glucose tolerance in men aged 59 to 70 years [22]
Thrifty phenotype hypothesis
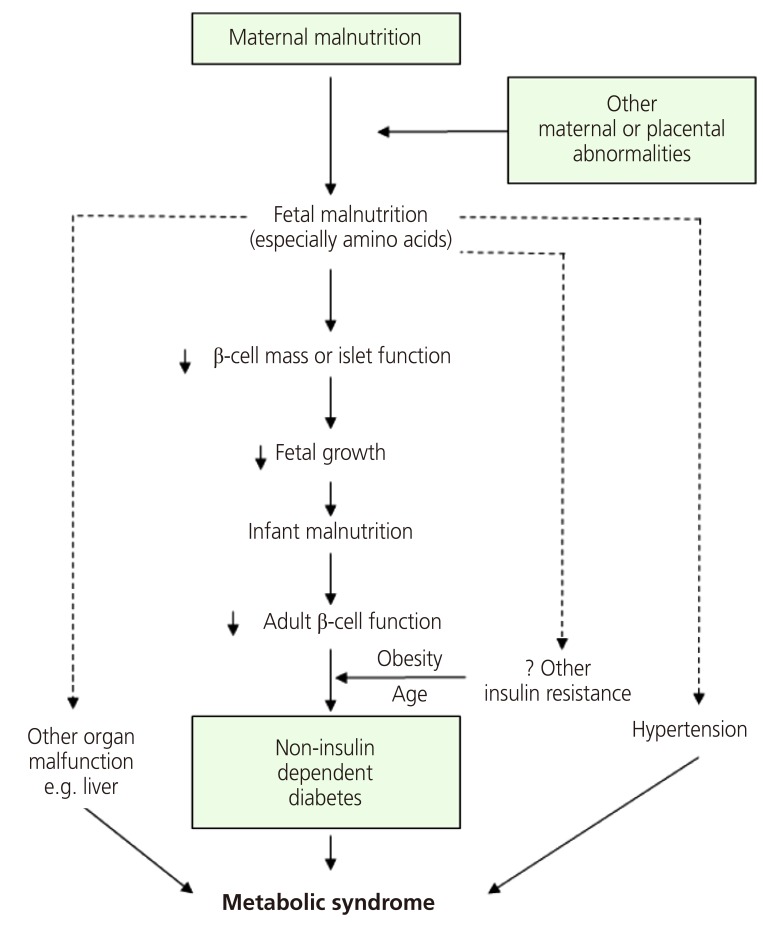
In a study of participants living in Hertfordshire, England, Hales et al. [22] found a correlation between weight and the prevalence of diabetes. They discovered that birth weight and weight at age 1 were inversely correlated with the prevalence of diabetes in adulthood and that the risk of IGT and diabetes mellitus fell with increasing birth weight. Consequently, they proposed a thrifty phenotype hypothesis for the etiology of type 2 diabetes, which asserts that malnutrition during the fetal and neonatal period causes hypoplasia of the endocrine pancreas and triggers hyposecretion of insulin by the pancreatic beta cells [24]. If a fetus is exposed to undernutrition or malnutrition during rapid cell differentiation of the pancreatic beta cells due to maternal malnutrition or maternal and placental abnormalities, it will try to overcome these limitations through metabolic programming. This process will cause impaired development of the endocrine pancreas, which results in poor insulin secretion. This alteration cannot be reversed even with subsequent adequate nourishment; as the fetus grows into adulthood, diabetes mellitus often develops when insulin requirements increase (Fig. 2). If malnutrition persists after birth, the insufficient insulin secretion is not harmful, and these subjects will simply show increased insulin sensitivity. However, calorie intake that leads to obesity, in which nutritional intake is increased and energy consumption is decreased, produces a physiological imbalance and results in glucose intolerance. As brain development is prioritized, organs such as the liver, vessels and pancreatic tissues are subject to various permanent structural and functional disorders. Additionally, the timing and combination of the nutritional deficiency determine the profile of metabolic disorders, such as diabetes mellitus, hypertension, dyslipidemia, and insulin resistance. Many studies have confirmed that fetal growth restriction and subsequent obesity aggravate glucose intolerance and insulin resistance [25]. In the thrifty phenotype hypothesis, “catch-up” growth is a critical factor for the manifestation of potential diseases in adulthood [26].
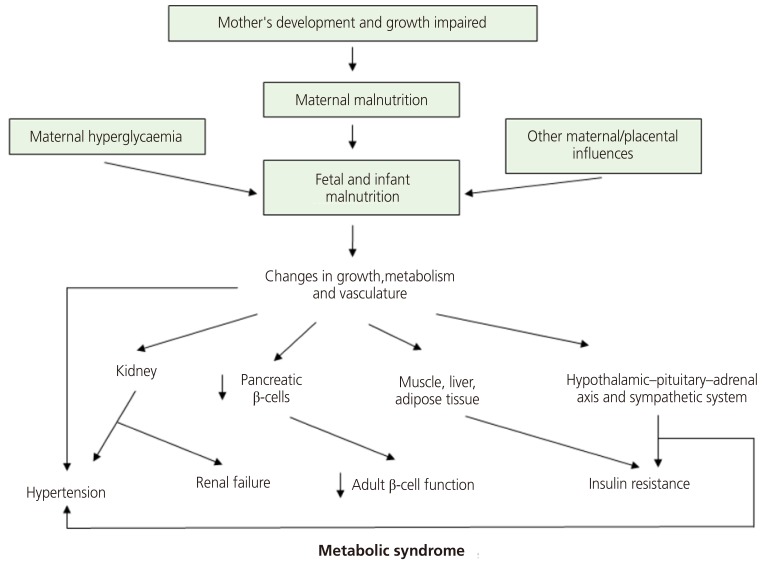
A recent update of this theory suggests maternal hyperglycemia is a risk factor for fetal nutritional deficiency. It also holds that changes in vessel structure and function, which are fundamental to the growth and functional modification of organs, is a consequence of fetal undernutrition. Finally, it incorporates the role of the hypothalamus-pituitary gland-adrenal gland (HPA) axis and the sympathetic nervous system (Fig. 3) [25].
Fig. 3
An updated diagram of the thrifty phenotype hypothesis incorporating recent findings and concepts. Also included are new speculative features; maternal hyperglycemia as predisposing factor and key roles of the vascular, hypothalamic-pituitary-adrenal axis and sympathetic systems [25].
Mechanism of fetal programming
Changes in endometrial endocrine, nutritional and cardiovascular conditions affect the expression of the fetal genome so it can adapt during development (Fig. 4). Within this framework, the fetal genome determines the intrauterine growth potential, but actual growth is primarily determined by environmental effects, such as fetal nutrition and the hormonal environment [27]. Fetal programming can affect individual gene expression at any stage, from changes in molecular biological functions, such as receptor cell density or sensitivity, to permanent hormonal changes or even alterations in metabolism or responses to physiological stressors [28].
Fig. 4
Fetal programming framework, indicating the possible role of the endocrine, nutrient, and cardiovascular milieu in utero [27].

1) Gene-imprinting (DNA methylation/chromatin remodeling)
2) Cell-alterations in the receptor density/metabolic breakdown of messengers
3) Organ-structural changes/alterations in organ volume/tissue composition
4) System-resetting of hormonal axes/altered stress responses
Nutrition delivered through the placenta is an especially important determinant of fetal growth because it allows the fetus to meet the growth potential determined by the underlying genotype. Recent reports have emphasized the impact of interactions between genotypes and the uterine environment. For example, siblings who share the same mother tend to have similar birth weights and show a 0.51 correlation, while siblings with the same father show a birth weight correlation of only 0.1. Other studies have indicated that cousins on the maternal side tend to have similar birth weights, whereas those on the paternal side do not. Babies born after oocyte donation have demonstrated that birth weight is strongly associated with the weight of the recipient rather than the weight of the donor [28]. One explanation for this may be related to the differential expression of the imprinted gene. Generally, imprinted genes expressed from the paternal system (IGF-2) stimulate growth, whereas imprinted genes (IGF-2 receptor) expressed from the maternal system act as growth inhibitors. The imprint of these genes is also controlled by epigenetic modifications, including DNA methylation and histone acetylation, under the control of environmental factors and nutrition [29].
1. Maternal glucocorticoid and cortisol secretion
The fetus is usually not significantly exposed to glucocorticoids until cortisol is produced by the adrenal gland near the end of pregnancy. The transfer of maternal glucocorticoids into the fetal circulation is restricted to the conversion of active cortisol to inactive cortisone by the placental enzyme 11β-hydroxysteroid dehydrogenase type-2 (11β-HSD2) [30]. In humans, degeneration of the 11β-HSD2 gene is associated with a low birth weight and reduced 11β-HSD2 activity, while increased fetal cortisol is correlated with delayed intrauterine growth [31,32]. In such a case, it is thought that there is significant exposure to maternal glucocorticoids, which can permanently change the state of the cardiovascular system, endocrine system and metabolism. Increased cortisol activity has been implicated in the pathogenesis of increased atherosclerosis, insulin resistance and cholesterol levels [33]. Cross-sectional studies have also suggested a relationship between low birth weight, increased cortisol levels and obesity. There is still some controversy regarding whether there is an effect on cortisol concentrations in the resting phase, but people with low birth weights have demonstrated exaggerated responses to stress and secreted significantly higher levels of cortisol than their normal birth weight counterparts [28].
The results of animal experiments after intrauterine exposure suggest that these changes in the HPA axis persist until adulthood [34,35]. For example, prenatal glucocorticoid exposure alters adult HPA function at every level of the axis from the brain to tissue glucocorticoid bioavailability [36]. These programmed changes in HPA function may contribute to metabolic disorders in adults. Tissue sensitivity to glucocorticoids would therefore increase as a result of reduced peripheral inactivation of cortisol in the fetus and altered expression of glucocorticoid receptors. This observation is also related to the clinical experience in which the fetus is exposed to steroids that travel through the placenta from glucocorticoids administered to the mother during preterm labor [37].
2. Kidney and liver function
Kidneys are exceptionally vulnerable to the effects of growth restriction. The easiest of these rates to identify are the kidney size and the number of kidney units (nephrons). This change is thought to be associated with fetal growth rate and supports the programmed body mass theory. Thereafter, any increase in growth and body weight rates into adulthood exceeds the functional retention of the kidneys, leading to an abnormal glomerular filtration rate, water accumulation and hypertension [38,39].
Data from Hertfordshire suggest that delayed intrauterine growth may alter liver function in adults by restricting the increase of LDL cholesterol and fibrinogen serum concentrations. Similarly, the same study demonstrated that a decrease in the circumference of the abdomen, which represents the size of the liver at birth, predicts the levels of serum cholesterol and fibrinogen in adulthood. These changes may be related to alterations in the way the liver circulation flows in the fetus [40,41].
3. Cardiovascular structure
A vascular reactivity study of low birth weight children showed decreased elasticity and a delayed vasodilation time post-stimulation. In animal and human studies, echocardiography revealed that growth was limited and structural heart defects were more common in offspring born with a low birth weight, including cardiomegaly [42]. This is likely the result of blood flow bypassing the peripheral circulation due to increased peripheral resistance. The initial pressure load affects cardiomyocyte development, leading to inadequate maturation in the early stages and resulting in fewer but larger myocytes. Evidence also suggests that this left ventricular hypertrophy may persist until adulthood [43,44].
4. Insulin resistance
Insulin resistance is an important component of metabolic syndrome and an important indicator of cardiovascular risk. Epidemiological studies have shown that both low birth weight children and adults have increased insulin resistance. Type 2 diabetes mellitus and insulin deficiency were observed to be the result of low birth weight. This relationship is supported by animal experiments that revealed correlations between a low birth rate and a decrease in the amount of pancreatic β-cells [45].
Insulin resistance is mainly seen at the periphery, and it has been suggested to be a result of adaptation to a limited supply of blood glucose to the central organs. For example, the prevalence of type 2 diabetes in sub-Saharan Africa is low, where fetal nutrition deficiencies and low birth weight as well as nutrition deficiency in childhood and adulthood are common. The migration of these rural people to the city has been associated with an increase in diabetes, again reaffirming the concept that programming to overcome short-term adversity in the womb is not beneficial in the long-term and may instead often be harmful later in life [46].
5. Growth hormone/insulin-like growth factor axis
The IGF family is known to play a crucial role in regulating fetal growth. For example, IGF-2 affects the growth of organs in embryos prior to implantation, while IGF-1 levels in umbilical cord blood have a direct relationship with birth weight [47]. Many studies have shown a link between cardiovascular disease, diabetes and the profile of osteoporosis and low serum levels of IGF-1. Nutritional deficiencies and the resulting associated growth restrictions are thought to lead to growth hormone resistance and further impairments in growth [48].
6. Adipose tissue, leptin, and insulin
Leptin is an adipocyte product that inhibits the production of insulin by affecting insulin gene transcription and secretion from pancreatic cells. Insulin also stimulates fat production. As fat storage increases, leptin production also rises to regulate the action of insulin. Animal studies indicate that the leptin-fat cell-insulin axis represents a direct link between body fat and the overproduction of insulin from fetal programming. Accordingly, leptin resistance is a contributor to obesity and hyperinsulinemia. Therefore, as obesity develops, leptin levels will increase but cannot neutralize hyperinsulinemia due to other effects [28].
The most important function of leptin is to lower appetite by acting on the hypothalamus. This explains the association between increased appetite, food intake and later obesity in an animal model of delayed intrauterine growth [49]. In the rat offspring of a 50% food-restricted mother, rapid catch-up growth significantly increased serum leptin levels and severe obesity at 6 months of age and was also observed along with gender differences [50]. In addition, umbilical cord concentrations of leptin are correlated with placental weight, body weight, length, and adiposity of human neonates [51].
7. Osteoporosis
It has been observed that changes in growth hormone and cortisol levels found after intrauterine growth restriction are related to an increased risk for osteoporotic fracture. The speculation is that intrauterine growth restriction alters growth plate sensitivity to growth hormones and cortisol. This perspective is supported by cohort studies where higher growth rates in young children are associated with increased risks of osteoporosis in adulthood [52].
The placenta's role in fetal programming
Several epidemiological studies have reported on the relationship between placental weight and the placental weight/birth weight ratio in fetal programming, and there are ongoing animal and human studies on the overall role of the placenta in fetal programming. The placenta regulates maternal-to-fetal nutritional composition and supply. It is also the source of hormonal signals that affect maternal and fetal metabolism. Proper development of the placenta is critical to normal fetal development and plays an active role in programming the in utero fetal experience, which influences diseases that may appear in adulthood. The function of the placenta develops gradually in a series of closely organized developmental stages of pregnancy. This sequential division may lead to abnormal development of placental vessels or trophoblasts [53]. The timing of any abnormalities in development will be crucial to the resulting placental function and thus to fetal programming. This damage, which alters placental development, includes hypoxia and abnormal maternal nutrition. In response, the placenta changes its transporter expression and activity to maintain fetal growth, resulting in epigenetic regulation [54] or placental gene expression [55]. Hypoxia is physiological in organogenesis, and placental tissue normally resides in a relatively hypoxic environment [56]; however, delayed intrauterine growth and preeclampsia are associated with a more severe hypoxia of trophoblasts. Metabolic activity of the placental mitochondria causes oxidative stress even in a normal pregnancy and is further intensified by intrauterine growth retardation, diabetes and preeclampsia [57]. It can also cause nitrative stress, which is known to bring about covalent modification and altered protein activity. Hypoxia, oxidation, and nitrification stress all alter placental development, and the associated changes in placental function may be the general fundamental mechanism underlying fetal programming (Fig. 5) [55].
Fig. 5
Placental adaptive responses and fetal programming [55].
11β-HSD, 11β-hydroxysteroid dehydrogenase; GLUT1, glucose transporter 1.

Gene expression
We observed genes that induced obesity and metabolic disorders in adults who were small for their gestational age due to nutritional deficiency (nutritional status) during pregnancy. In the study, when the offspring of a pregnant mouse given only 50% of the normal amount of food had its brain dissected at 3 weeks of age, a significant difference was observed compared to the control in the expressed levels of ubiquitin carboxylase terminal hydrolase L1 and secretin (Fig. 6) [58]. Methylenetetrahydrofolate dehydrogenase 1 and S-methyl transferase 1, enzymes related to single carbon metabolism in the livers of this animal model, were also lower than in the control, which was associated with high levels of blood homocysteine (Fig. 7). As blood homocysteine levels rise, there is an increased health risk of cardiovascular diseases and Alzheimer's disease [59]. In addition, wide spaces between hepatic cells were induced in mice on a low nutrition diet compared to normal mice (Fig. 8A), and a dramatic increase in gene expression levels of the mammalian target of rapamycin and sterol regulatory element-binding protein 1 were also observed (Fig. 8B and C). Thus, these results suggest that the liver of small-for-gestational-age newborns of mice that were exposed to a low nutrition diet showed dysfunctional liver signaling and lipid metabolism and had higher incidences of insulin resistance, diabetes and non-alcoholic fatty liver disease as adults [60]. Another piece of evidence suggests that when the pregnant mice were fed a low-protein diet, the liver of the offspring of the mother showed increased expression of glucocorticoids and paroxysmal proliferator-activated receptor, which is related to metabolic function. Such gene-specific changes may influence the occurrence of hypertension and hyperlipidemia later in life [61].
Fig. 6
The secernin-1 (SCRN1) and ubiquitin carboxy-terminal hydrolase L1 (UCHL1) protein expression in the whole brain of 3-week-old offspring by using Western blot analysis [58].
C, control; FR, food-restricted.
All values are given as mean±standard deviation. a)
P<0.05; b)
P<0.01 (Wilcoxon rank-sum test).

Fig. 7
Comparison of liver protein expression in the offspring according to maternal diet [59]. The ratio of betaine-homocysteine methyltransferase 1 (BHMT1), methylenetetrahydrofolate dehydrogenase 1 (MTHFD1), and ATP synthase subunit beta, mitochondrial (ATP5B) density in the livers of male (A) and female (B) offspring. The number of samples (S) signify the protein density of the offspring in each group. The levels of expression of BHMT1, MTHFD1, and ATP5B were significantly reduced in the livers of the food-restricted (FR)/ad libitum (AdLib) male offspring (P<0.05, C) compared with those of the control group. The levels of BHMT1, MTHFD1, and ATP5B in the livers of female offspring did not differ among groups (D). Data are expressed as mean±standard deviation. a)P<0.05 compared with AdLib/AdLib; b),c)Significant difference of the protein expressions between sampe protein (P<0.05).

Fig. 8
A photomicrograph of male offspring livers in the 3 groups (A); the protein expression levels of mammalian target of rapamycin (mTOR) and sterol regulatory element-binding protein 1 (SREBP1) were measured by Western blotting in the 3 groups [60].
AdLib/FR, given an ad libitum diet during late gestation and an 50% food-restricted diet during lactation; cv, central vein; FR/AdLib, given a 50% food-restricted diet during late gestation and ad libitum diet during lactation.
Data are present as mean±standard deviation. P-values indicate the significance of the differences among the groups (one-way analysis of variance). a)Control vs. FR/AdLib, FR/AdLib vs. AdLib/FR, control vs. AdLib/FR, P<0.05.

Conclusion
Cardiovascular disease and type 2 diabetes are 2 of the leading causes of death worldwide. During the last century, an increase in the migration of rural populations to urban areas has led to significant dietary changes and an increased degree of obesity. In evolutionary terms, fetal programming is not intended to help adults survive longer but rather exists to provide a mechanism for the fetus to adapt to conditions in the intrauterine environment. Fetal programming may predispose people to certain adult diseases and is an important matter of concern in the current age of low fertility and an aging population because it suggests that the aggressive treatment of low birth weights during pregnancy can prevent many adult diseases. However, the mechanism for the fetal programming of adulthood diseases remains a hypothesis, and there is insufficient understanding of the mechanism and also no method to determine if a fetus has been predisposed to certain adult diseases. Therefore, it is necessary to look beyond the epidemiological relevance to understand and study the mechanisms at the cellular and molecular level. Nutritional studies have focused on macro-nutrients in the past. Current investigations of micro-nutrients and also consideration of programming in the fetal environment due to factors other than nutrition are therefore required.
Epidemiological studies on possible fetal origins of adult diseases have only been conducted in adults; further studies on disease susceptibility in childhood and also from birth to adulthood will be required. Mutual strategic studies that include clinical and epidemiological studies as well as laboratory experiments should additionally be carried out. Currently, the health of the fetus is considered more important than ever due to the trends in low birth rates and late childbearing; understanding the role and mechanism of fetal programming, which has a negative impact on future health as well as on fetal health, has important academic value for finding intervention strategies.






")











